Running the Workflows
NMDC EDGE Quick Start User Guide

Register for an account
Users must register for an account within the NMDC EDGE platform or login using the user’s ORCiD account.

User Profile
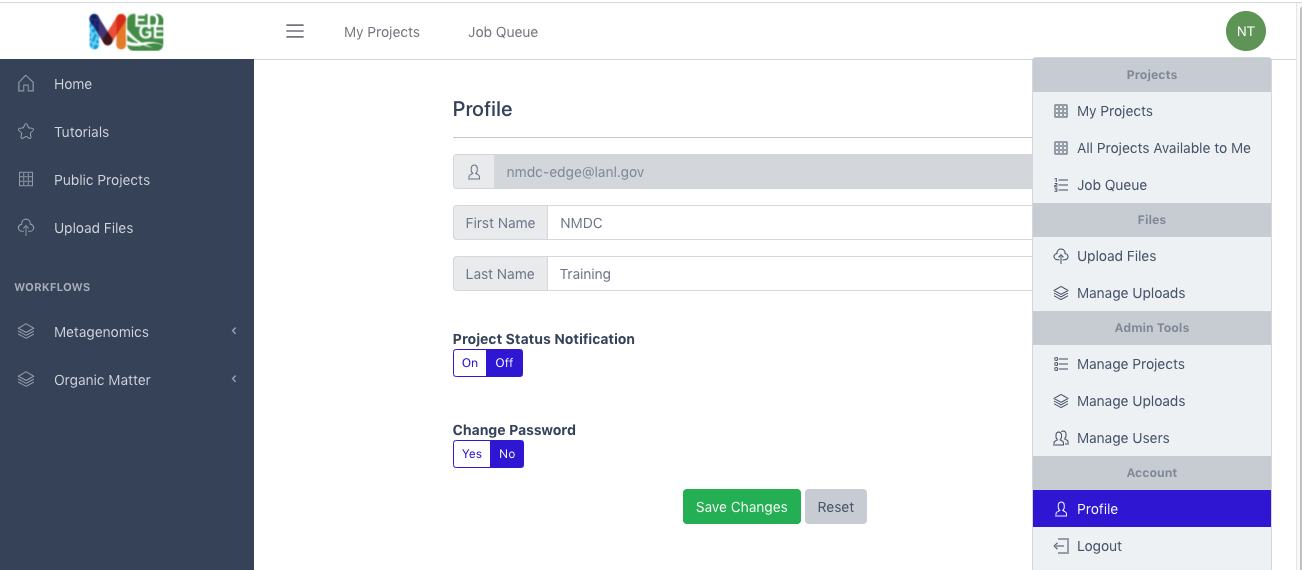
Once logged in, the green button with the user’s initials on the right provides a drop-down menu which allows the user to manage their projects and uploads; there is also a button which allows users to edit their profile. On this profile page, there are two options: 1) the option to receive email notification of a project’s status (OFF by default) and 2) the option to change the user’s password (also OFF by default).
Upload data
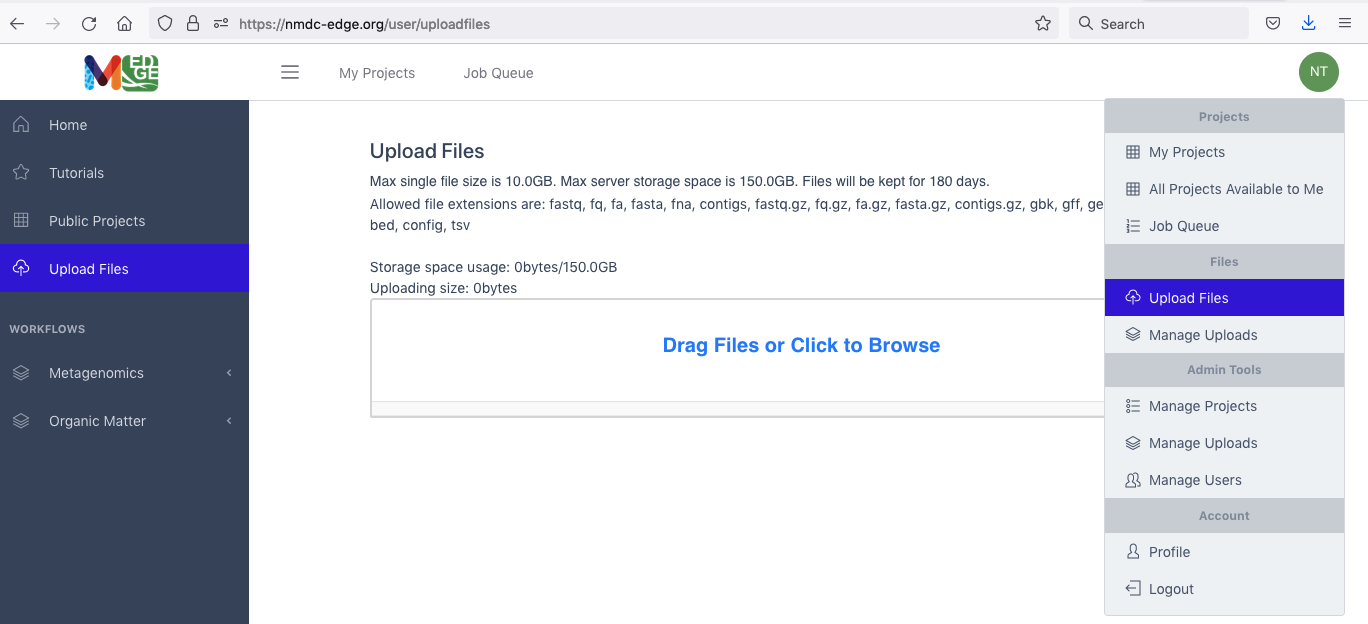
Two options are available for users to upload their own data to process through the workflows. The first is using the button in the left menu bar. The second is through the drop-down menu shown when clicking the green button with the user’s initials on the right. Either button will open a window which allows the user to drag and drop files or browse for the user’s data files. (There are also some datasets in the Public Data folder for users to test the platform.)
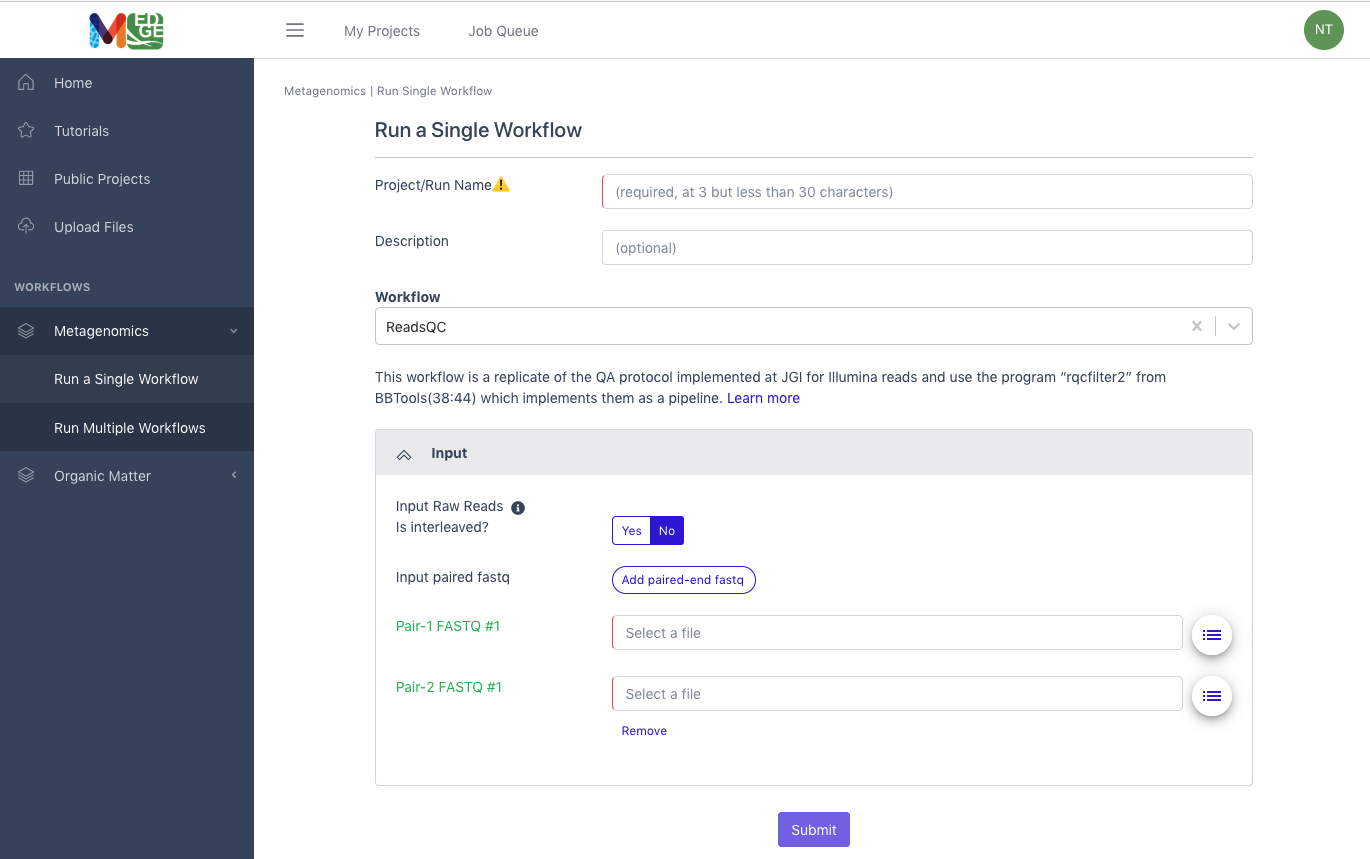
Running a single workflow
To run a workflow, the user must provide:
A unique Project/Run Name with no spaces (underscores are fine).
A description is optional, but recommended.
The user then selects the workflow desired from the drop-down menu.
For metagenomic/metatranscriptomic data, the user must also select if the input data is interleaved or separate files for the paired reads.
Then the input file(s) from the available list of files.
The user should click “Submit.
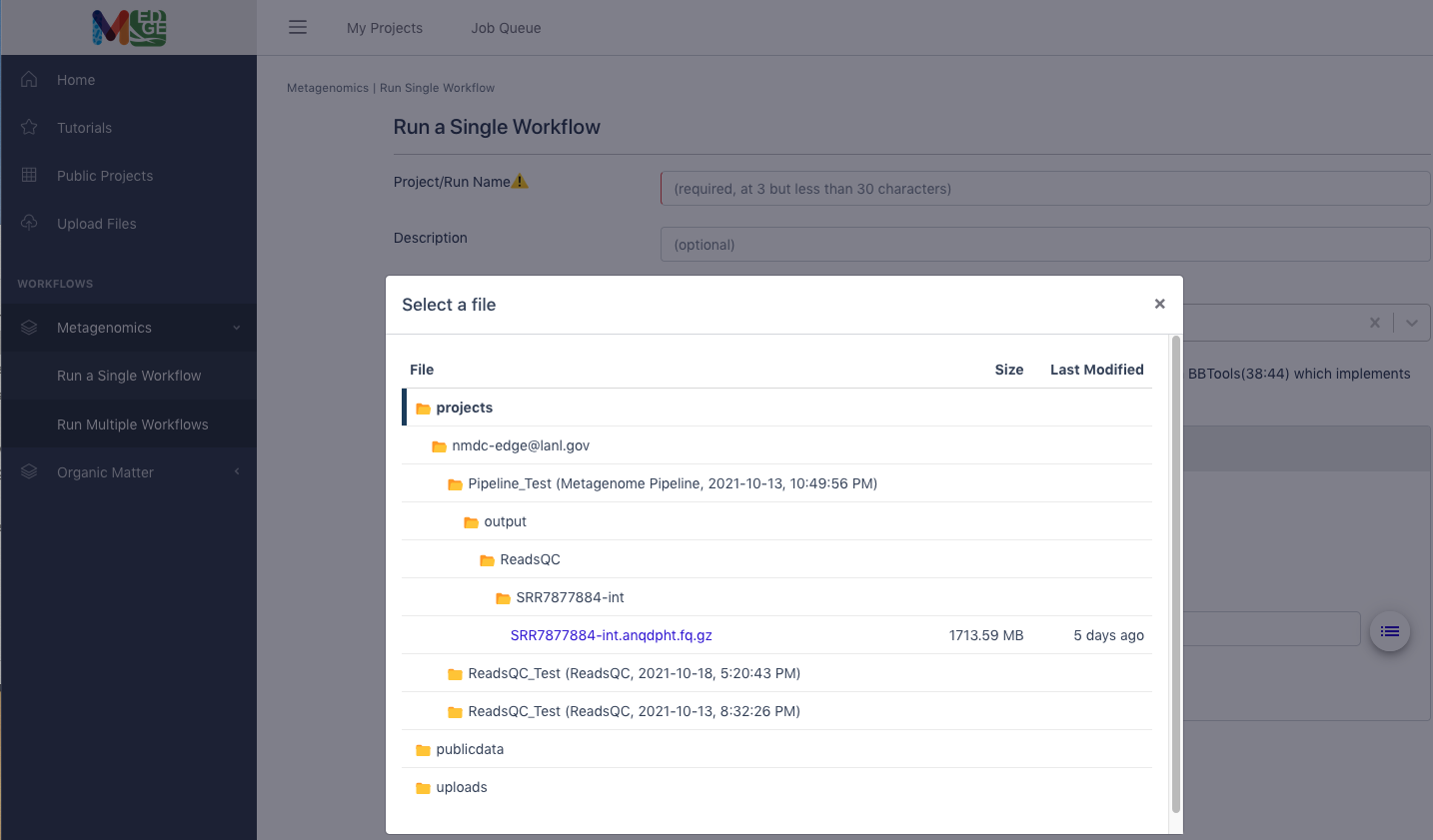
Note: Clicking on the buttons to the right of the data input blanks opens a box called “Select a file” to allow the user to find the desired files (shown in purple) from previously run projects, the public data folder, or user uploaded files.
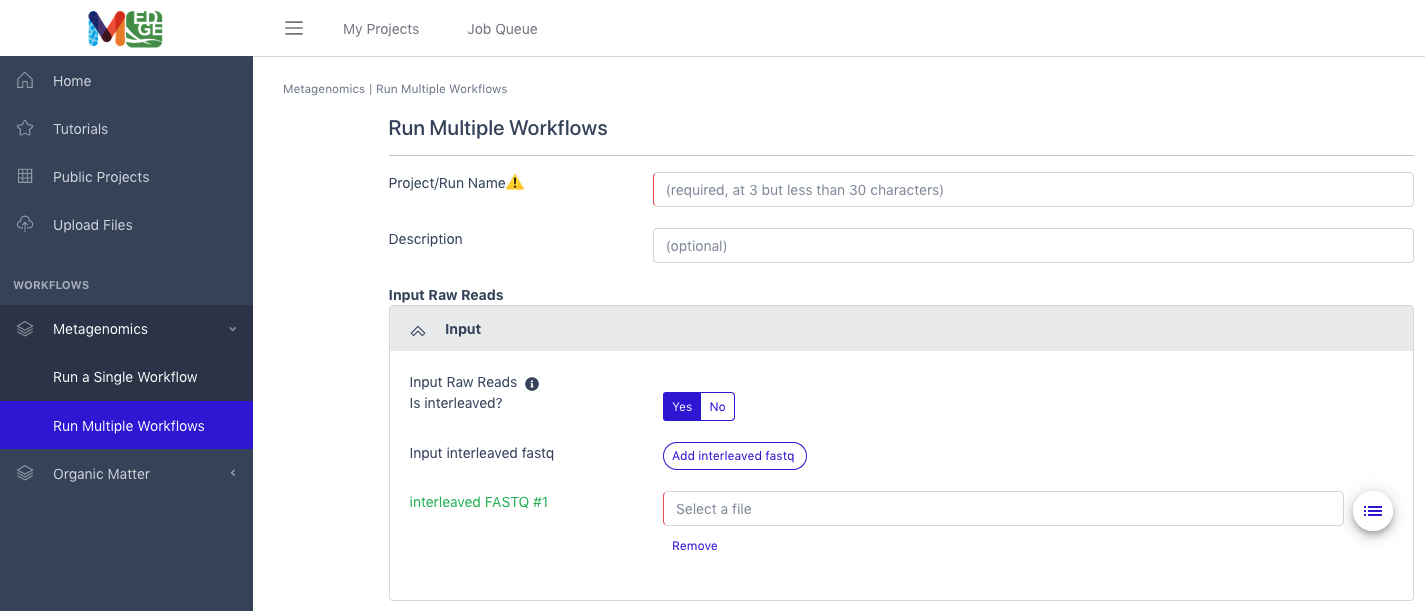
Running multiple workflows
Another option is to select “Run Multiple Workflows” if the user desires to run more than one of the metagenomic workflows or the entire metagenomic pipeline.
Enter a unique Project/Run Name with no spaces (underscores are fine).
A description is optional, but recommended.
The user must also select if the input data is interleaved or separate files for the paired reads.
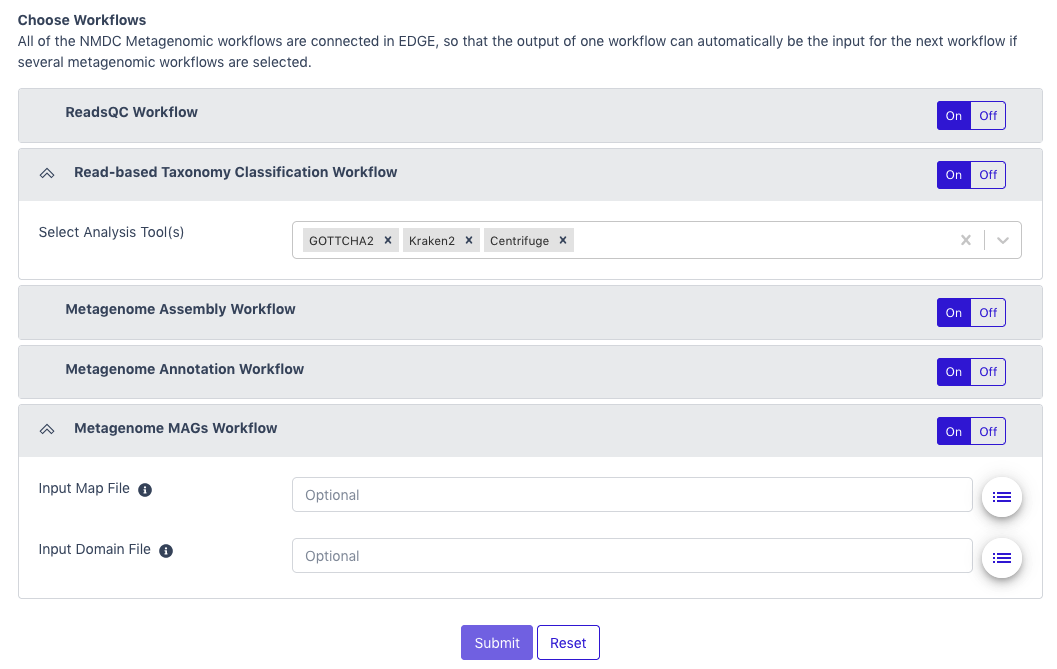
All five of the metagenomic workflows are “ON” by default, but the user
can select to turn off any workflows not desired. The pipeline uses the
output of each workflow as the input for subsequent workflows. (Note:
Some workflows require input data from prior workflows, so turning one
workflow off may result in other workflows also automatically turning
off.) Then the user can click “Submit.”
Output
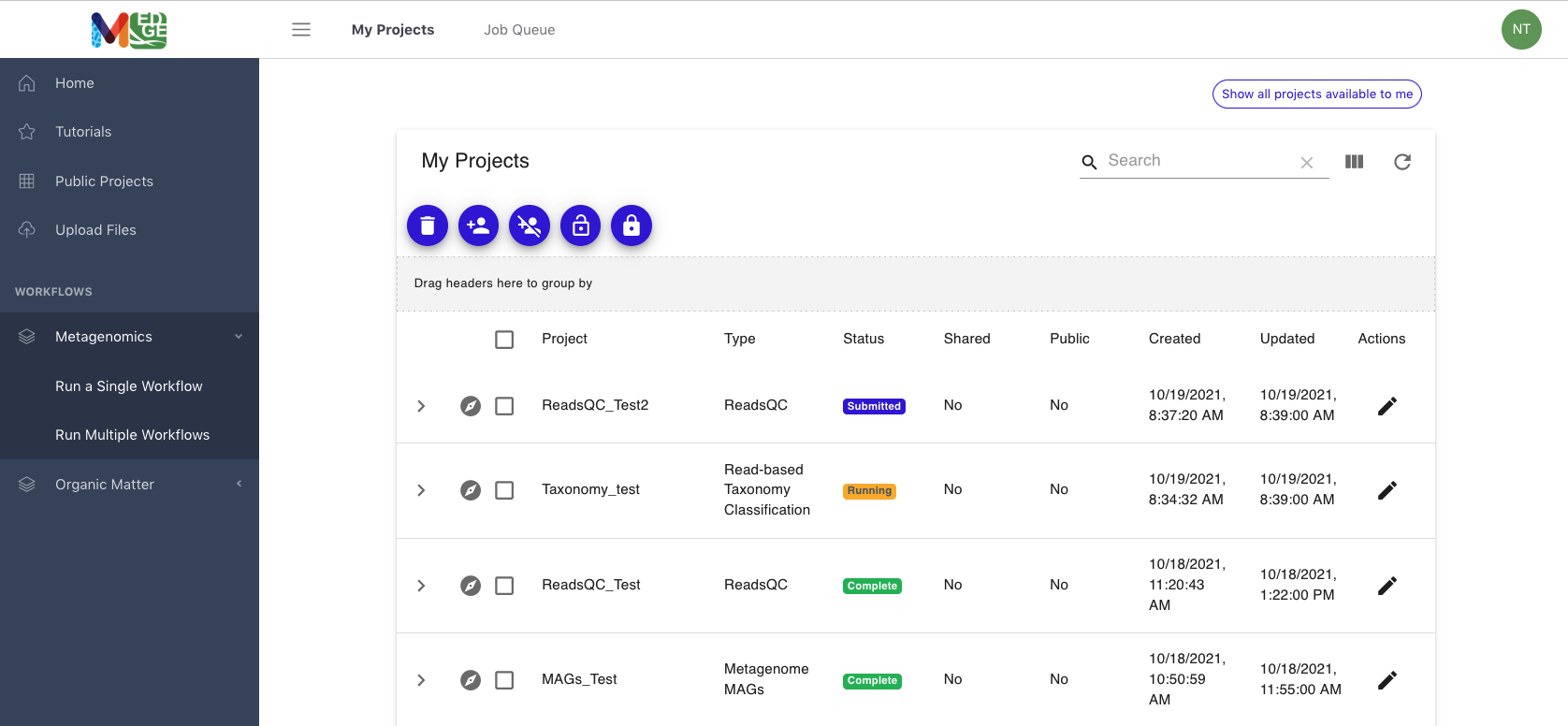
The link for ‘My Projects’ opens the list of projects for that user
Links (in the purple circles) are provided to share projects, make projects public, or delete projects
The “Status” column shows whether the job is in the queue (gray), submitted (purple), running (yellow), has failed (red) or completed (green). If a project fails, a log will give the error messages for troubleshooting.
Clicking on the icon to the left of a project name opens up the results page for that project.
Project Summary (Results)
The project summary page will show three categories. Clicking on the bar or tab opens up the information.
General contains the project run information.
“Workflow” Result contains the tabular/visual output.
Browser/Download Outputs contains all the output files available for downloading. There may be several folders.
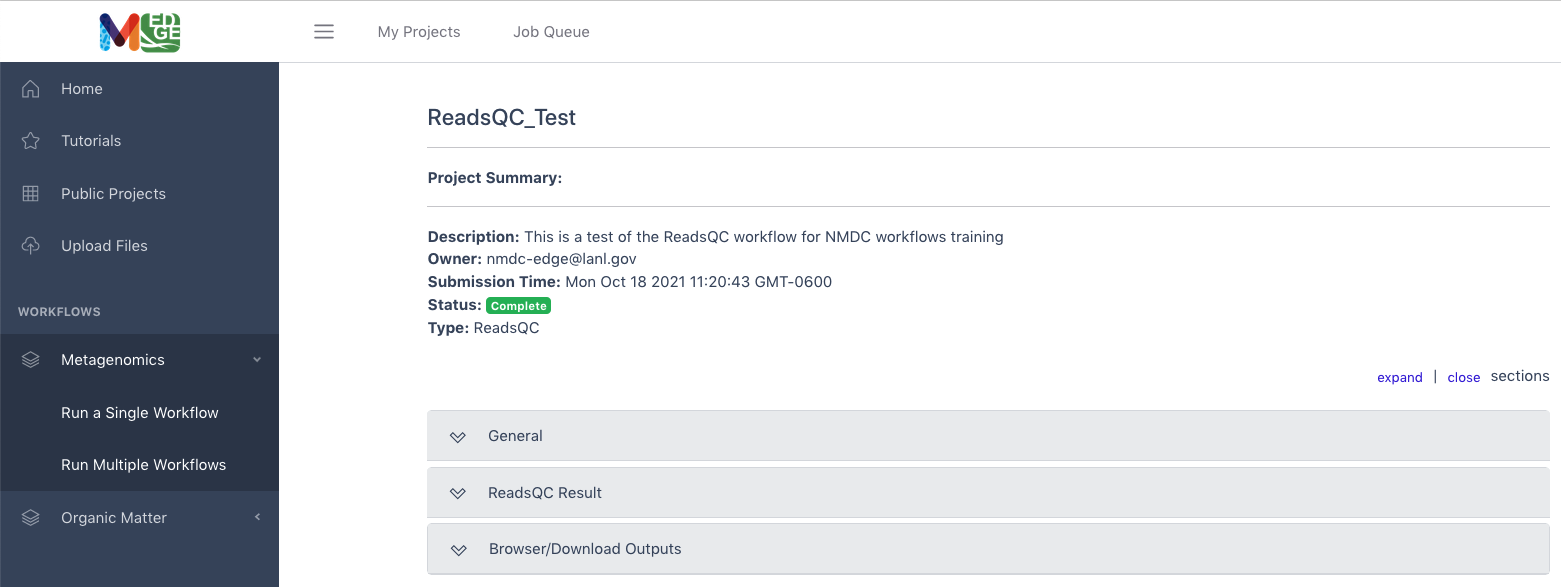
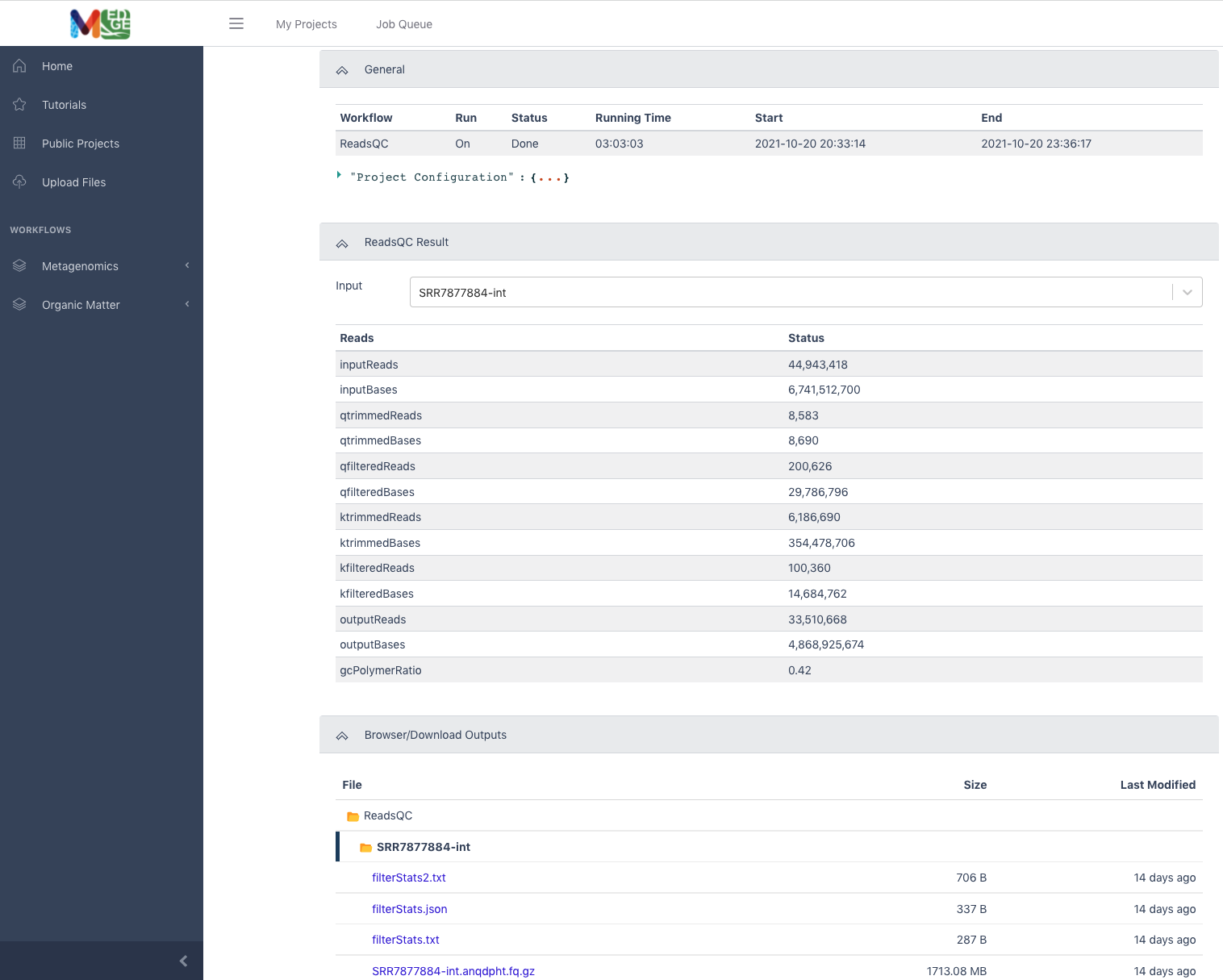
This example shows the results of a ReadsQC workflow run which shows run time under the General tab, the workflow results of quality trimming and filtering under the ReadsQC Results tab, and the files available for download (shown in purple) under the Browser/Download Outputs tab.
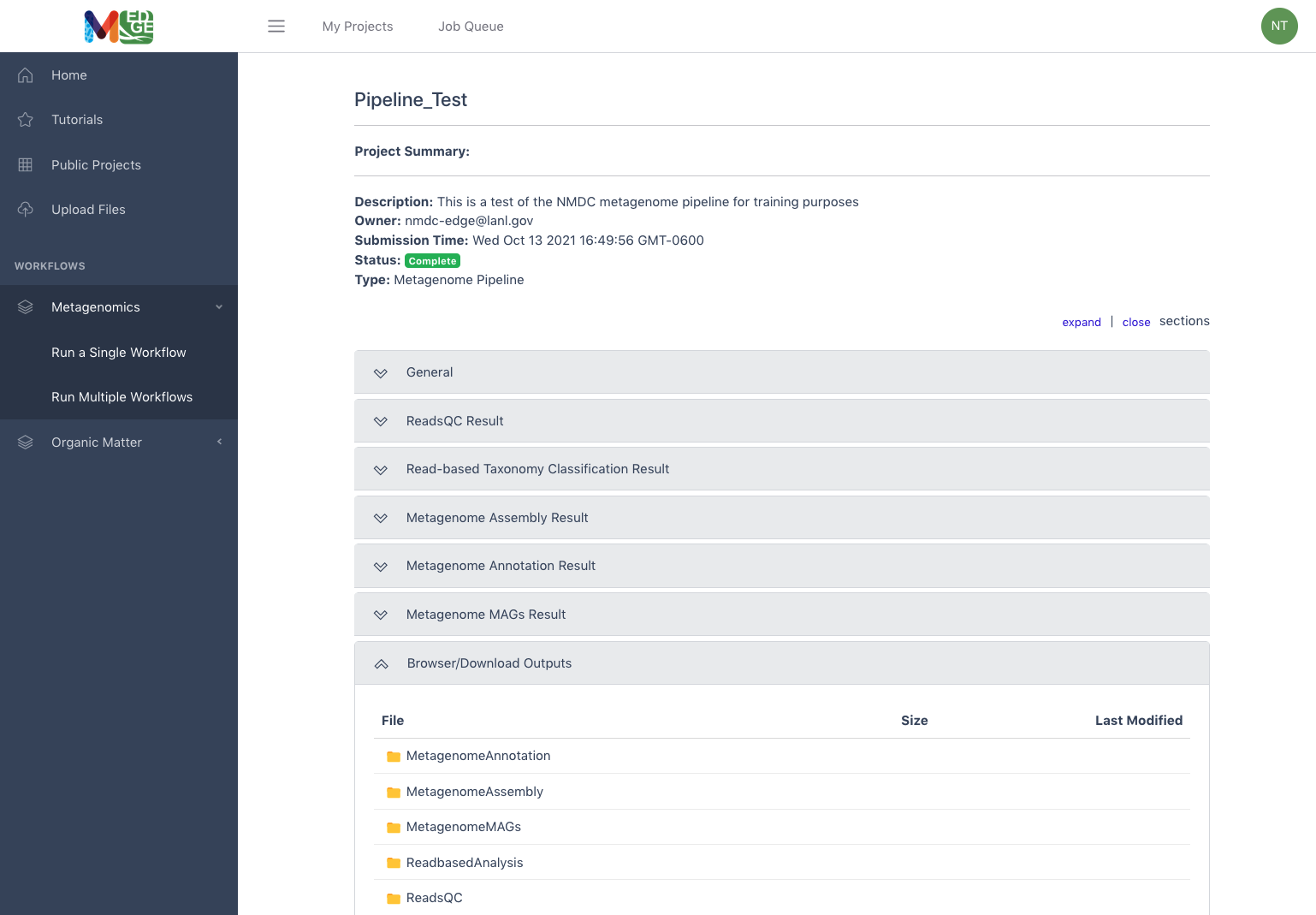
The full Metagenome pipeline or “Multiple Workflow” run results show the results of each workflow under a separate tab and the associated files available for download are in separate workflow folders under the Browser/Download Outputs tab.
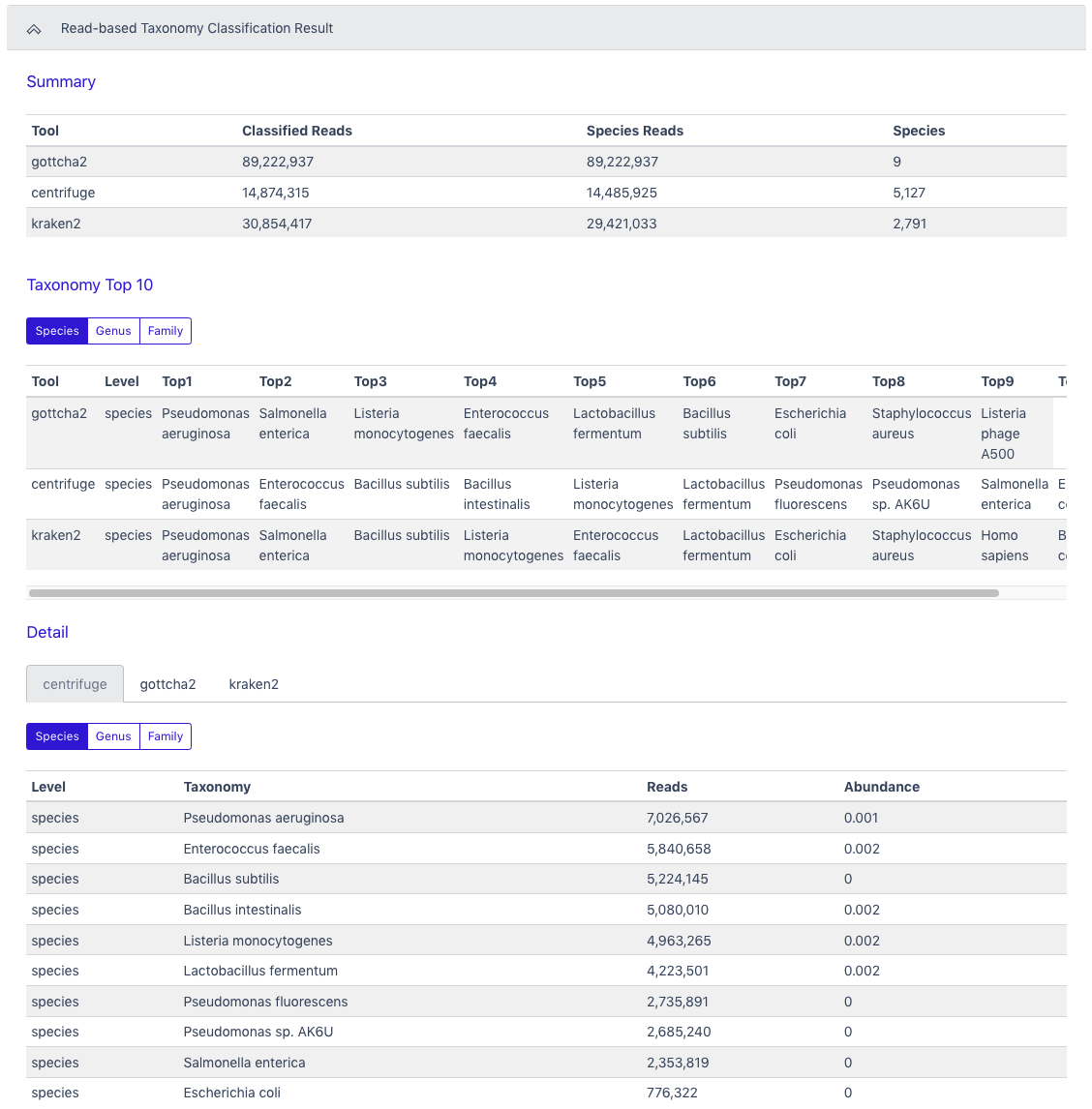
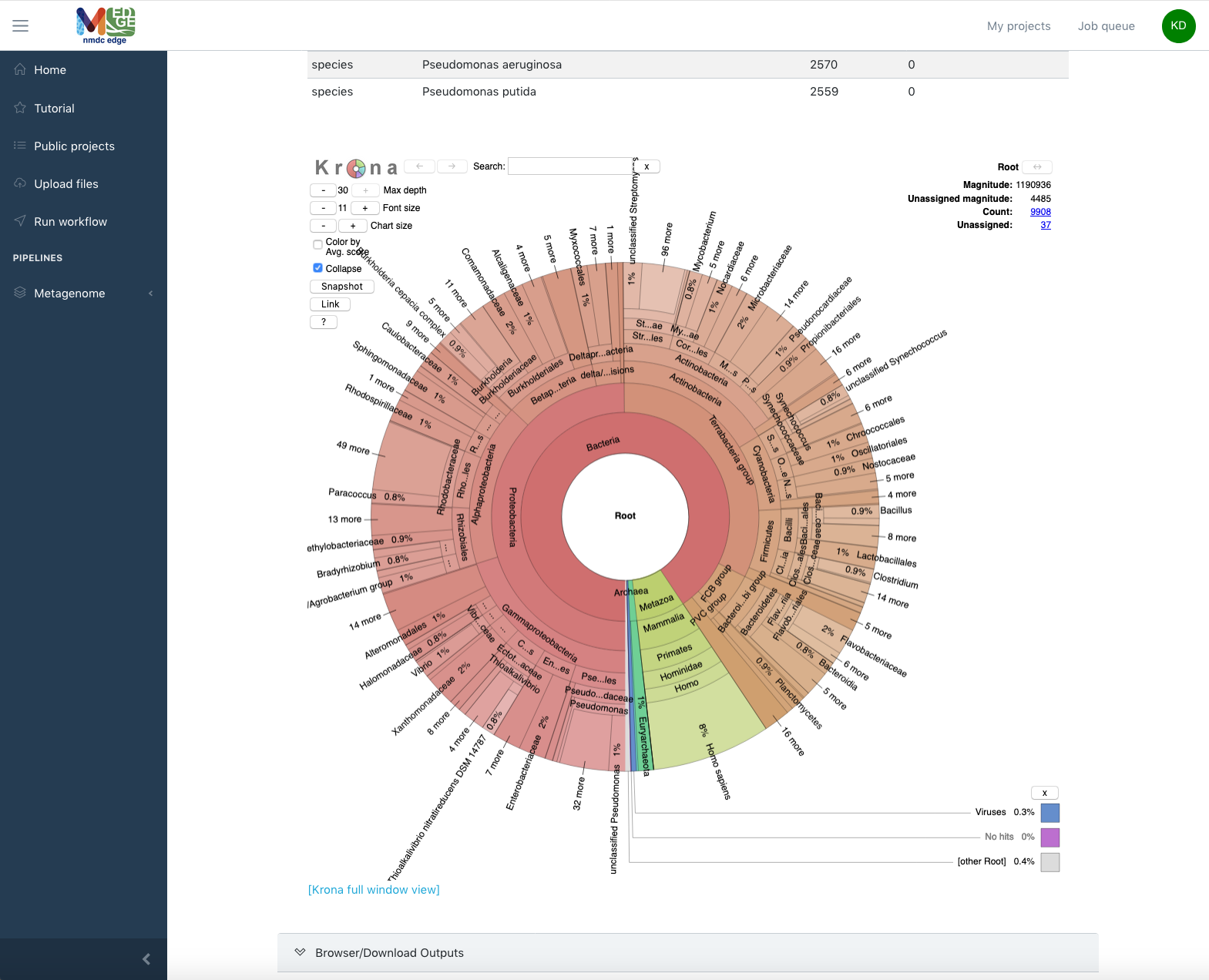
As a second example, the next two figures show the results from the Read-based Taxonomy Classification workflow. The summary includes classified reads and the number of species identified for all of the selected taxonomy classifiers. The top ten organisms identified by each tool at three taxonomic levels is also provided. Tabs for each of the classification tools providing more in-depth results are in the Detail section. Krona plots are generated for the results at each of the three taxonomic levels for each of the tools and can also be found in the Detail section. Full results files (beyond the Top 10) and the graphics are available for download.
Metagenomics Workflows
ReadsQC
Overview
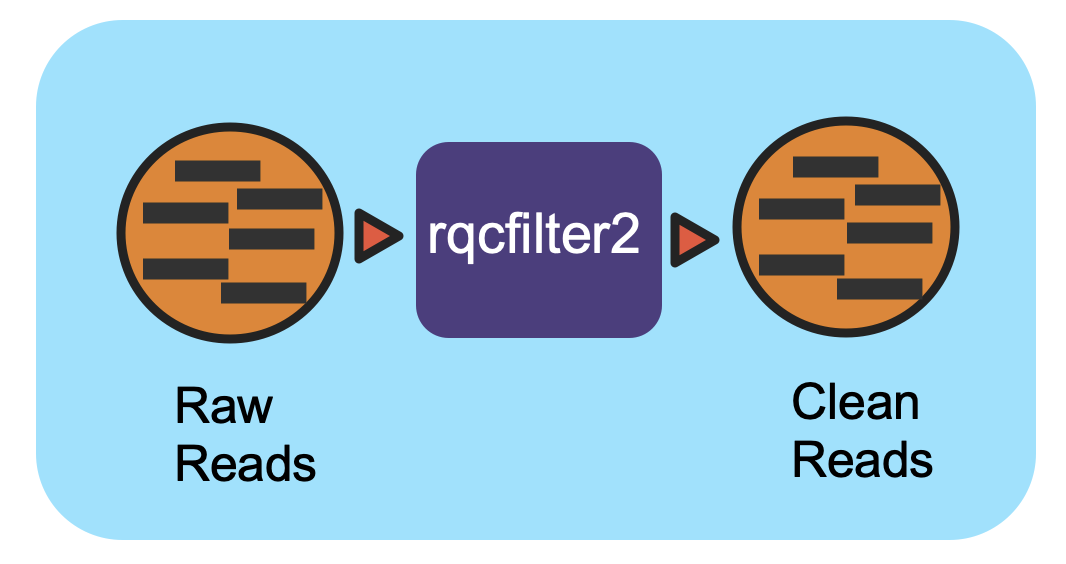
This workflow performs quality control on raw Illumina reads to trim/filter low quality data and to remove artifacts, linkers, adapters, spike-in reads and reads mapping to several hosts and common microbial contaminants.
Running the Workflow
Currently, this workflow is available in GitHub and can be run from the command line. (CLI instructions and requirements are found here.) Alternatively, this workflow can be run in NMDC EDGE.
Input
Metagenome ReadsQC requires paired-end Illumina data as an interleaved file or as separate pairs of FASTQ files.
Acceptable file formats: .fastq, .fq, .fastq.gz, .fq.gz
Details
This workflow performs quality control on raw Illumina reads using rqcfilter2. The workflow performs quality trimming, artifact removal, linker trimming, adapter trimming, and spike-in removal using bbduk, and performs human/cat/dog/mouse/microbe removal using bbmap. Full documentation can be found in ReadtheDocs.
Software Versions
rqcfilter2 (BBTools v38.94)
bbduk (BBTools v38.94)
bbmap (BBTools v38.94)
Output
Multiple output files are provided by the workflow; the primary files are shown below. The full list of output files can be found in ReadtheDocs.
| Primary Output Files | Description |
|---|---|
| Filtered Sequencing Reads | Cleaned paired-end data in interleaved format (.fastq.gz) |
| QC statistics (2 files) | Reads QC summary statistics (.txt) |
Running the Reads QC Workflow in NMDC EDGE
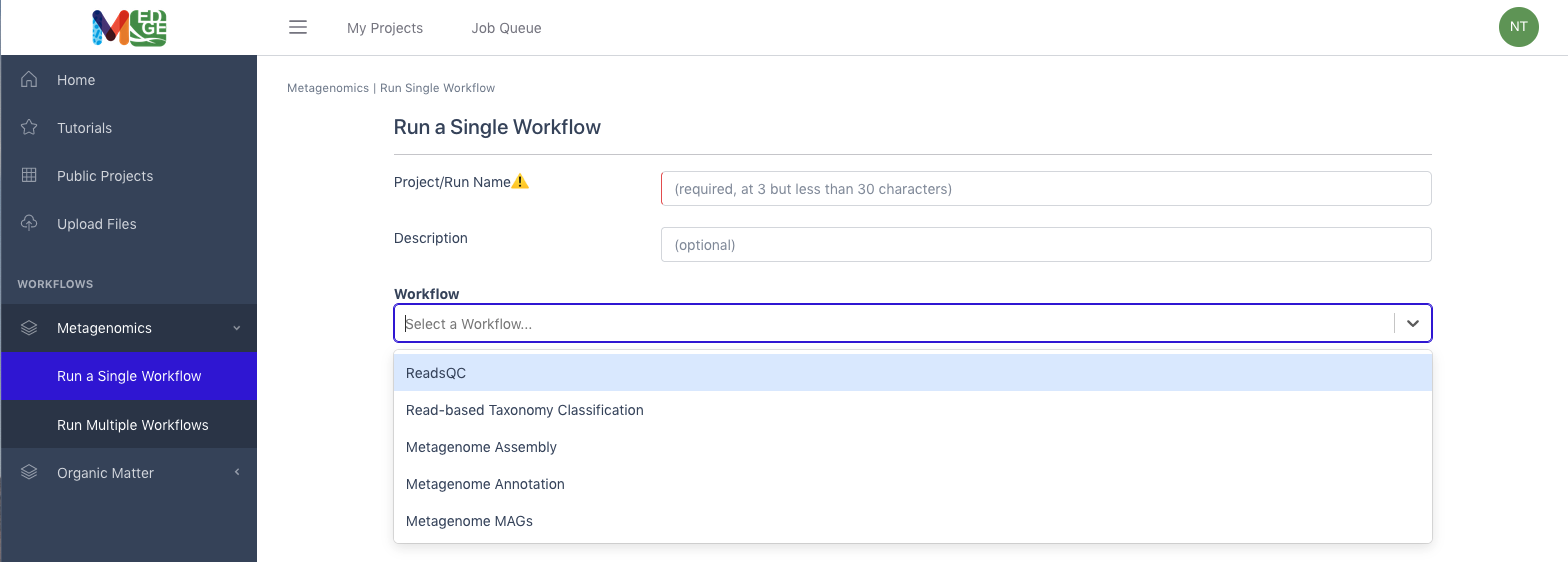
Select a workflow
From the Metagenomics category in the left menu bar, select ‘Run a Single Workflow’.
Enter a unique project name with no spaces (underscores are fine).
A description is optional, but helpful.
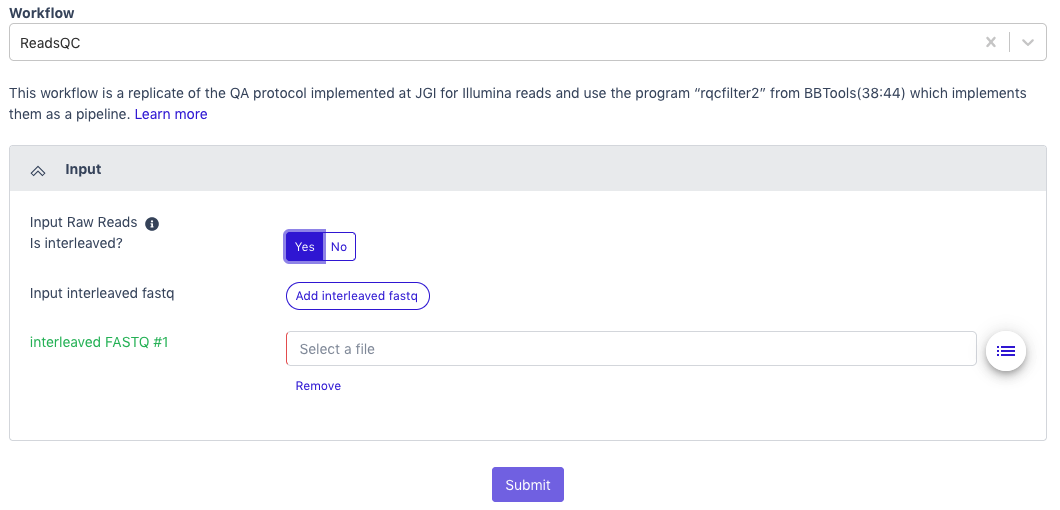
Select ‘ReadsQC’ from the dropdown menu under Workflow.
Input
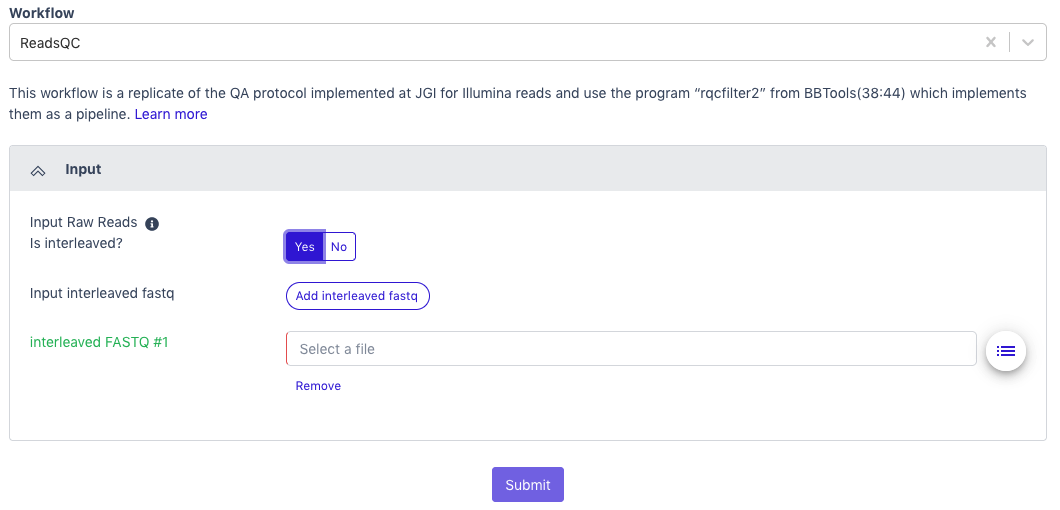
ReadsQC requires paired-end Illumina data in FASTQ format as the input; the file can be interleaved and can be compressed. Acceptable file formats: .fastq, .fq, .fastq.gz, .fq.gz
The default setting is for the raw data to be in an interleaved format (paired reads interleaved into one file). If the raw data is paired reads in separate files (forward and reverse), click ‘No’.
Additional data files (of the same type–interleaved or separate) can be added with the button below.
Click the button to the right of the input blank for data to select the data file for the analysis. (If there are separate files, there will be two input blanks.) A box called ‘Select a File’ will open to allow the user to find the desired file(s) from previously run projects, the public data folder, or files uploaded by the user.
Then click ‘Submit’.
Output
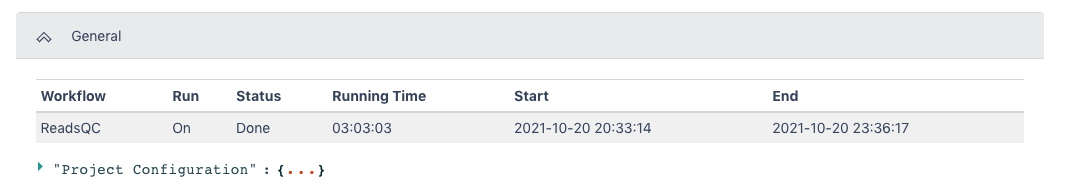
The General section of the output shows which workflow was run and the run time information.
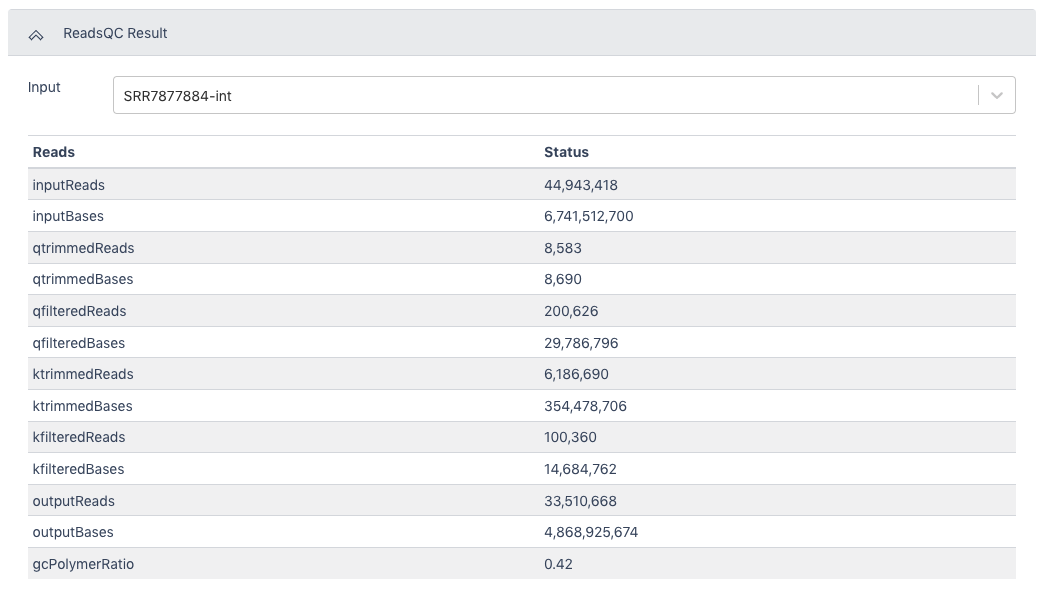
The ReadsQC Result section shows the data input and provides a variety of metrics including the number of reads and bases before and after trimming and filtering.
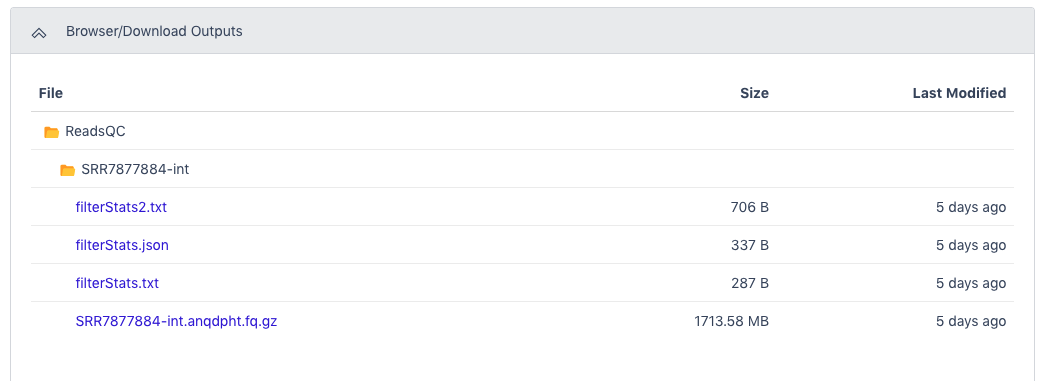
The Browser/Download Output section provides output files available to download. The clean data will be in an interleaved .fq.gz file. General QC statistics are in the filterStats.txt file.
Read-based Taxonomy Classification
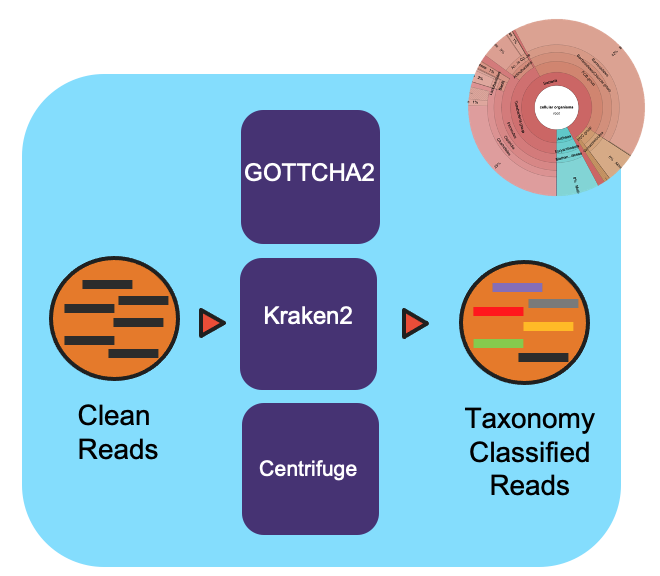
Overview
This workflow takes in Illumina sequencing files (single-end or paired-end) and profiles the reads using multiple taxonomic classification tools.
Running the Workflow
Currently, this workflow is available in GitHub and can be run from the command line. (CLI instructions and requirements are found here.) Alternatively, this workflow can be run in NMDC EDGE.
Input
The Metagenome Read-based Taxonomy Classification workflow requires Illumina data and can accept data as an interleaved file or as separate pairs of FASTQ files. Interleaved data will be treated as single-end reads. (It is highly recommended to input clean data from the ReadsQC workflow.)
Acceptable file formats: .fastq, .fq, .fastq.gz, .fq.gz
Details
To create a community profile, this workflow utilizes three taxonomy classification tools: GOTTCHA2, Kraken2, and Centrifuge. These tools vary in levels of specificity and sensitivity. Each tool has a separate reference database. These databases (152 GB) are built into NMDC EDGE. Users can select one, two, or all three of the classification tools to run in the workflow. Full documentation can be found in ReadtheDocs.
Software Versions
GOTTCHA2 v2.1.6
Kraken2 v2.0.8
Centrifuge v1.0.4
Output
Multiple output files are provided by the workflow; the primary files are shown below. The full list of output files can be found in ReadtheDocs.
| Primary Output Files | Description |
|---|---|
| Profiling results for each tool | Tabular results of the profile for each tool (.tsv) |
| Krona plots for each tool | Interactive graphic file (.html) |
Running the Read-based Taxonomy Classification Workflow in NMDC EDGE
Select a workflow
From the Metagenomics category in the left menu bar, select ‘Run a Single Workflow’.
Enter a *unique project name with no spaces (underscores are fine).
A description is optional, but helpful.
Select ‘Read-based Taxonomy Classification’ from the dropdown menu under Workflow.
Input
This workflow accepts Illumina data in FASTQ format as the input; the file can be interleaved and can be compressed. This input can be the output from the ReadsQC workflow and this is recommended. Acceptable file formats: .fastq, .fq, .fastq.gz, .fq.gz
The default setting is for the raw data to be in an interleaved format (paired reads interleaved into one file). If the raw data is paired reads in separate files (forward and reverse), click ‘No’.
Additional data files (of the same type–interleaved or separate) can be added with the button below.
Click the button to the right of the input blank for data to select the data file for the analysis. (If there are separate files, there will be two input blanks.) A box called ‘Select a File’ will open to allow the user to find the desired file(s) from previously run projects, the public data folder, or files uploaded by the user.
Then click ‘Submit’.
Output
The General section of the output shows which workflow and which tools were run and the run time information.
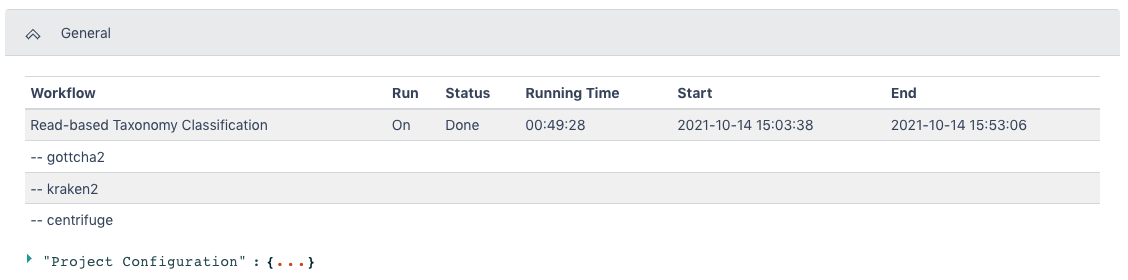
The Read-based Taxonomy Classification Result section has a summary section at the top and results for each tool at three levels of taxonomy in the Taxonomy Top 10 section. The Detail section has classified reads results and relative abundance results for each tool at three levels of taxonomy.
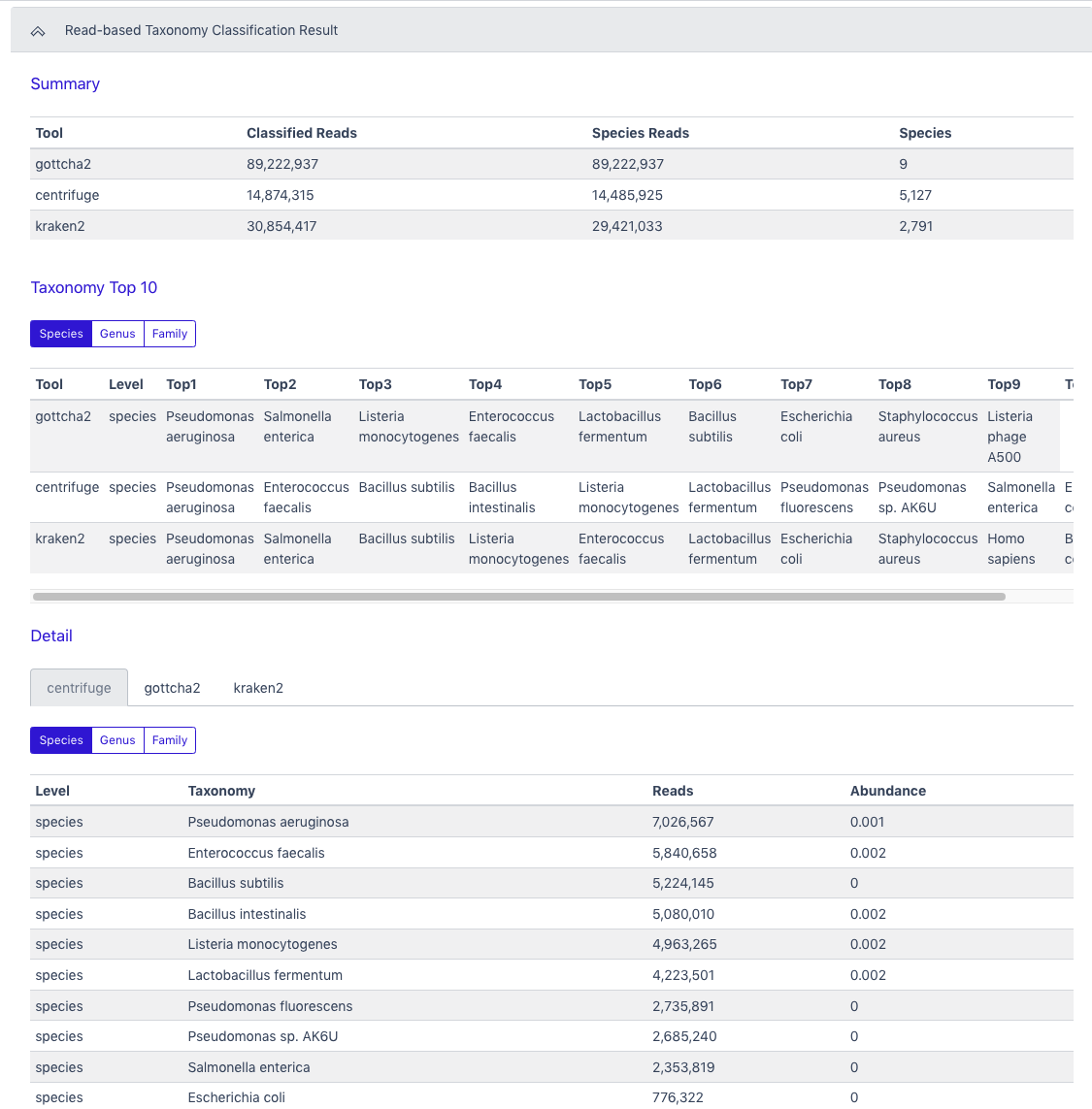
The Detail section also provides an interactive Krona plot for each tool.
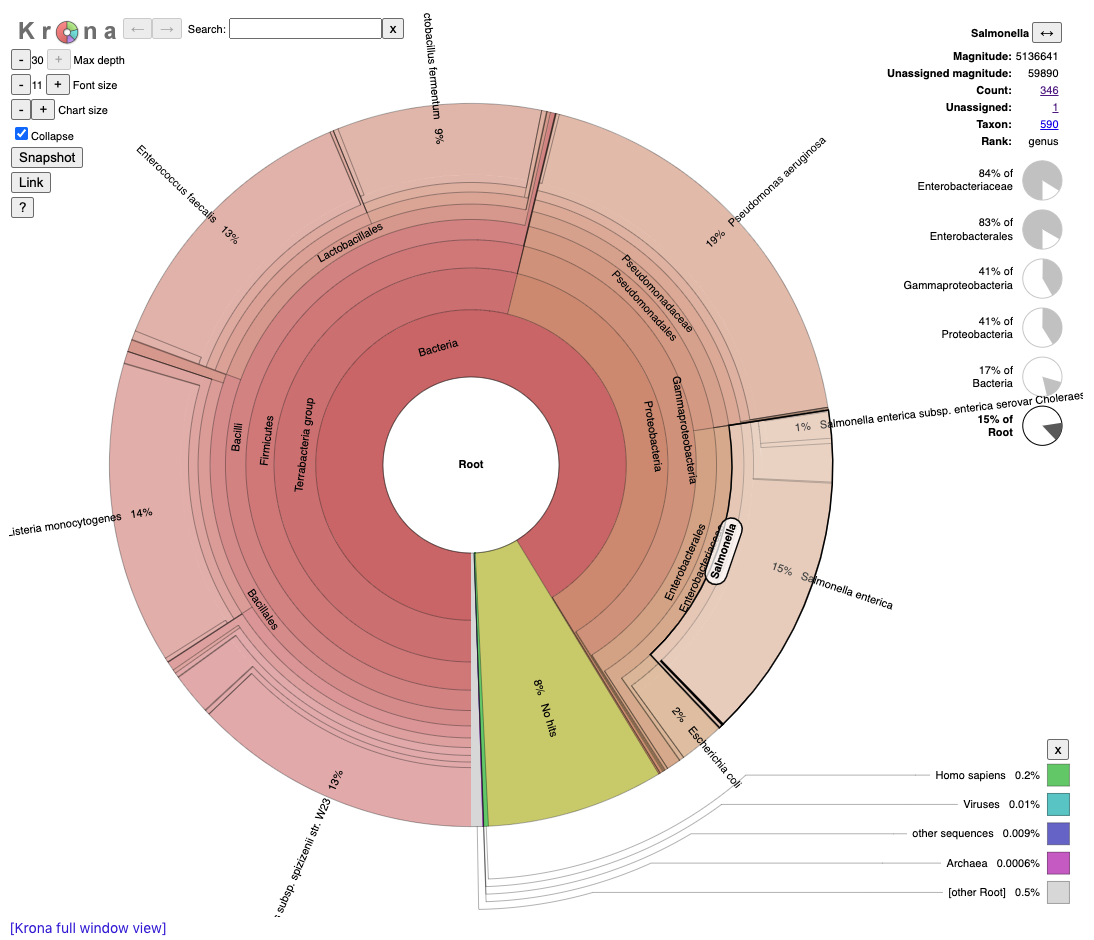
The Browser/Download Output section provides output files available to download. Each tool has a separate folder for the results from that tool. Full tabular results are in the largest .tsv file and the interactive Krona plots (.html files) open in a separate browser window.
Assembly
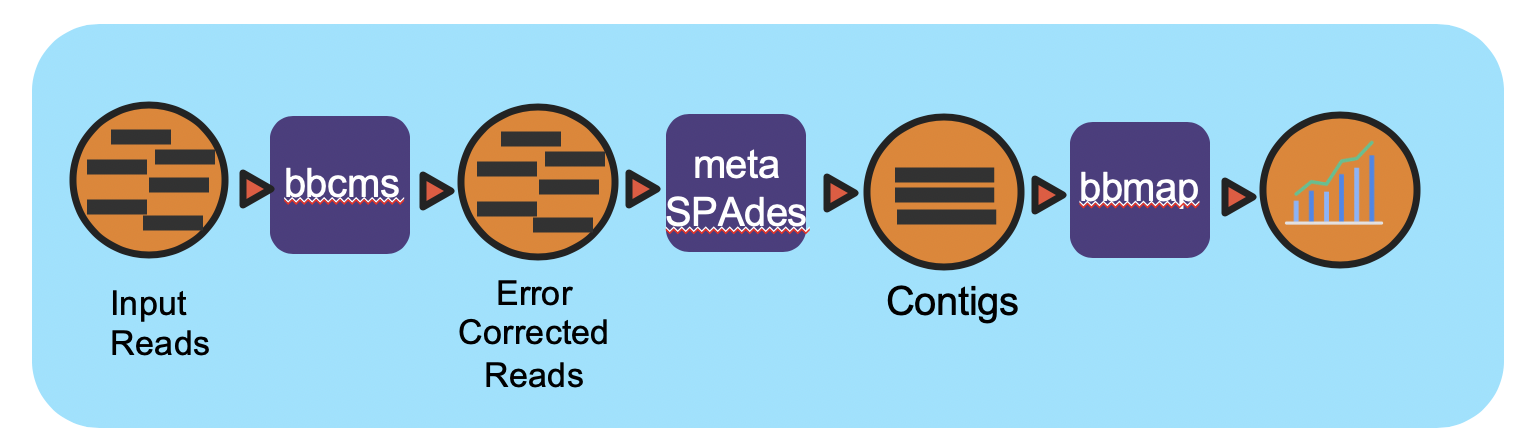
Overview
This workflow takes in paired-end Illumina data, runs error correction, assembly, and assembly validation.
Running the Workflow
Currently, this workflow is available in GitHub and can be run from the command line. (CLI instructions and requirements are found here.) Alternatively, this workflow can be run in NMDC EDGE.
Input
Metagenome Assembly requires paired-end Illumina data as an interleaved file or as separate pairs of FASTQ files. The recommended input is the output from the ReadsQC workflow.
Acceptable file formats: .fastq, .fq, .fastq.gz, .fq.gz
Details
This workflow takes in paired-end Illumina reads and performs error correction using bbcms. Then the corrected reads are assembled using metaSPAdes. After assembly, the reads are mapped back to the contigs using bbmap for coverage information. Full documentation can be found in ReadtheDocs.
Software Versions and Parameters
bbcms (BBTools v38.94)
metaSpades v3.15.0
bbmap (BBTools v38.94)
Output
Multiple output files are provided by the workflow; the primary files are shown below. The full list of output files can be found in ReadtheDocs.
| Primary Output Files | Description |
|---|---|
| Assembly Contigs | Final assembly contigs (assembly_contigs.fna) |
| Assembly Scaffolds | Final assembly scaffolds (assembly_scaffolds.fna) |
| Assembly AGP | An AGP format file which describes the assembly |
| Assembly Coverage BAM | Sorted bam file of reads mapping back to the final assembly |
| Assembly Coverage Stats | Assembled contigs coverage information |
Running the Metagenome Assembly Workflow in NMDC EDGE
Select a workflow
From the Metagenomics category in the left menu bar, select ‘Run a Single Workflow’.
Enter a unique project name with no spaces (underscores are fine).
A description is optional, but helpful.
Select ‘Metagenome Assembly’ from the dropdown menu under Workflow.

Input
This workflow accepts Illumina data in FASTQ format as the input; the file can be interleaved and can be compressed. (It is highly recommended to input clean data from the ReadsQC workflow.)
Acceptable file formats: .fastq, .fq, .fastq.gz, .fq.gz
The default setting is for the raw data to be in an interleaved format (paired reads interleaved into one file). If the raw data is paired reads in separate files (forward and reverse), click ‘No’.
Additional data files (of the same type–interleaved or separate) can be added with the button below.
Click the button to the right of the input blank for data to select the data file for the analysis. (If there are separate files, there will be two input blanks.) A box called ‘Select a File’ will open to allow the user to find the desired file(s) from previously run projects, the public data folder, or files uploaded by the user.
Then click ‘Submit’.
Output
The General section of the output shows which workflow was run and the run time information.
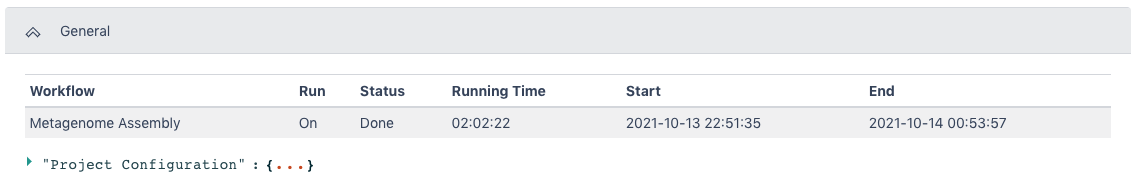
The Metagenome Assembly Result section has all of the statistics from the assembly.
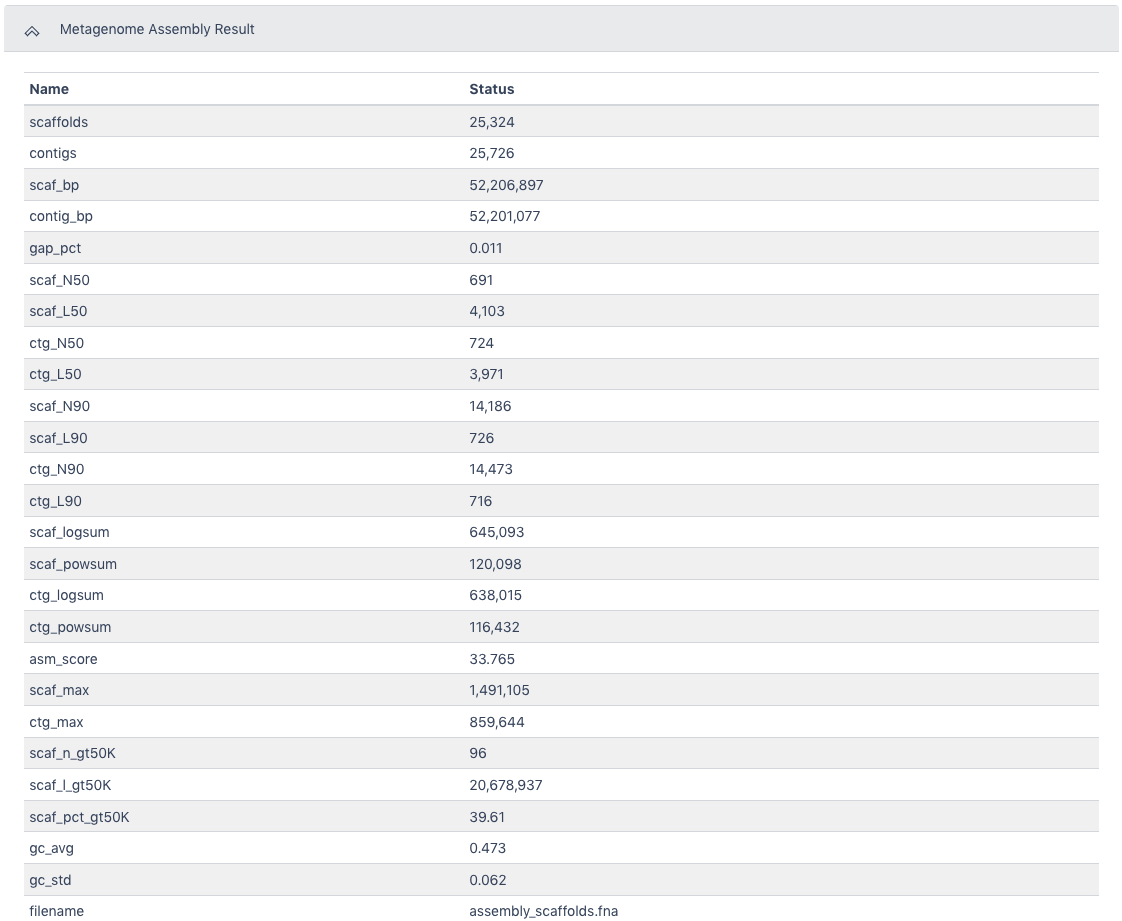
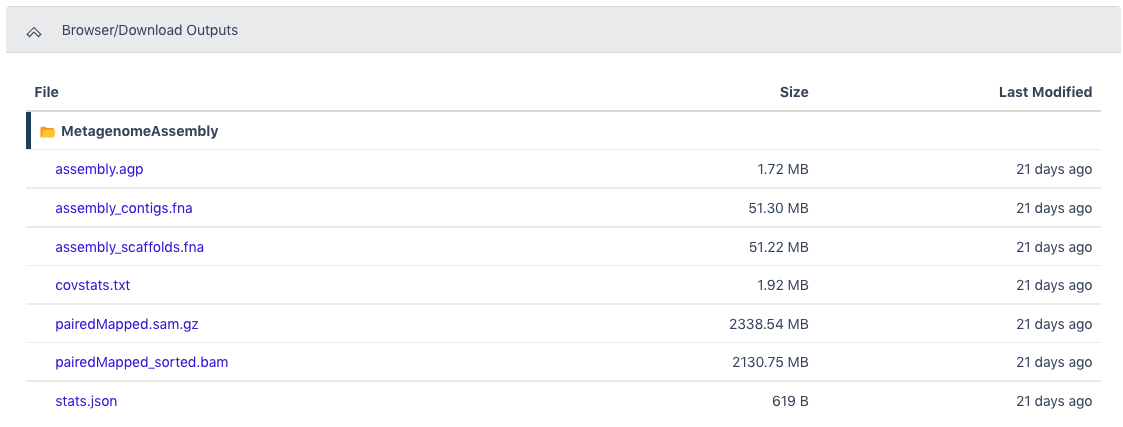
The Browser/Download Output section provides output files available to download. The primary result is the assembly_contigs.fna file which can also be the input for the Metagenome Annotation workflow. The pairedMapped_sorted.bam file along with the assembled contigs file can be the input for the MAGs Generation workflow.
Annotation
Overview
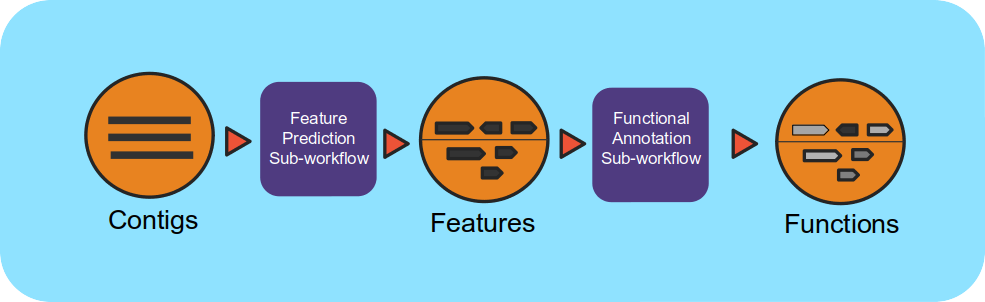
This workflow takes assembled metagenomes and generates structural and functional annotations.
Running the Workflow
Currently, this workflow is available in GitHub and can be run from the command line. (CLI instructions and requirements are found here.) Alternatively, this workflow can be run in NMDC EDGE.
Input
Metagenome Annotation requires assembled contigs in a FASTA file. This input can be the output from the Metagenome Assembly workflow and this is recommended.
Acceptable file formats: .fasta, .fa, .fna, .fasta.gz, .fa.gz, .fna.gz
Details
The workflow uses a number of open-source tools and databases to generate the structural and functional annotations. The input assembly is first split into 10MB splits to be processed in parallel. Depending on the workflow engine configuration, the split can be processed in parallel. Each split is first structurally annotated, then those results are used for the functional annotation. The structural annotation uses tRNAscan_se, RFAM, CRT, Prodigal and GeneMarkS. These results are merged to create a consensus structural annotation. The resulting GFF is the input for functional annotation which uses multiple protein family databases (SMART, COG, TIGRFAM, SUPERFAMILY, Pfam and Cath-FunFam) along with custom HMM models. The functional predictions are created using Last and HMM. These annotations are also merged into a consensus GFF file. Finally, the respective split annotations are merged together to generate a single structural annotation file and single functional annotation file. In addition, several summary files are generated in TSV format. Full documentation can be found in ReadtheDocs.
Software Versions
Conda
tRNAscan-SE >= 2.0
Infernal 1.1.2
CRT-CLI 1.8
Prodigal 2.6.3
GeneMarkS-2 >= 1.07
Last >= 983
HMMER 3.1b2
TMHMM 2.0
Output
Multiple output files are provided by the workflow; the primary files are shown below. The full list of output files can be found in ReadtheDocs.
| Primary Output Files | Description |
|---|---|
| Structural Annotation | Consensus structural annotation file from multiple tools (.gff) |
| Functional Annotation | Consensus functional annotation file from multiple tools (.gff) |
| KEGG summary | KEGG gene function tabular summary (.tsv) |
| EC summary | Enzyme Commission tabular summary (.tsv) |
| Gene phylogeny summary | Gene phylogeny tabular summary (.tsv) |
Running the Metagenome Annotation Workflow in NMDC EDGE
Select a workflow
From the Metagenomics category in the left menu bar, select ‘Run a Single Workflow’.
Enter a unique project name with no spaces (underscores are fine).
A description is optional, but helpful.
Select ‘Metagenome Annotation’ from the dropdown menu under Workflow.
Input
This workflow accepts assembled Illumina data in FASTA format as the input; the file can be compressed. (It is highly recommended to input the assembled contigs from the Metagenome Assembly workflow.) Acceptable file formats: .fasta, .fa, .fna, .fasta.gz, .fa.gz, .fna.gz.
Click the button to the right of the input blank for data to select the data file for the analysis. (If there are separate files, there will be two input blanks.) A box called ‘Select a File’ will open to allow the user to find the desired file(s) from previously run projects, the public data folder, or files uploaded by the user.
Then click ‘Submit’.
Output
The General section of the output shows which workflow was run and the run time information.
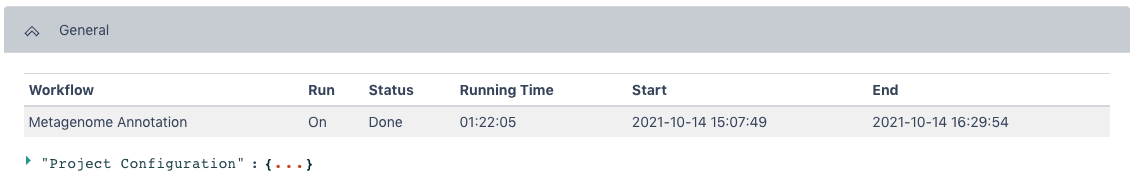
The Metagenome Annotation Result section has statistics for Processed Sequences, Predicted Genes, and General Quality Information from the workflow.
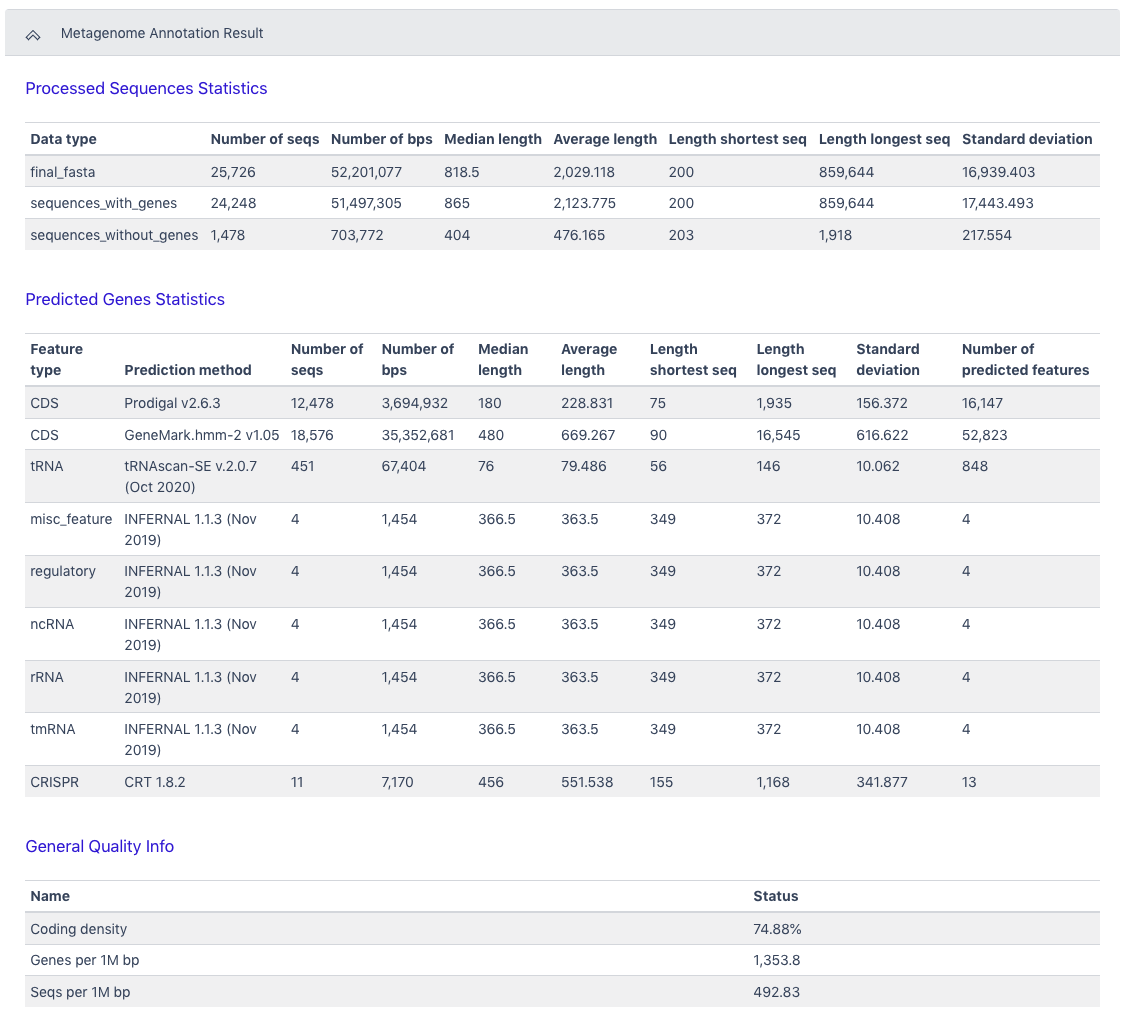
The Browser/Download Output section provides output files available to download. The primary results are the functional annotation and the structural annotation files (.gff). The functional annotation file is required input for the MAGs Generation workflow along with the assembled contigs.
MAGs Generation
Overview
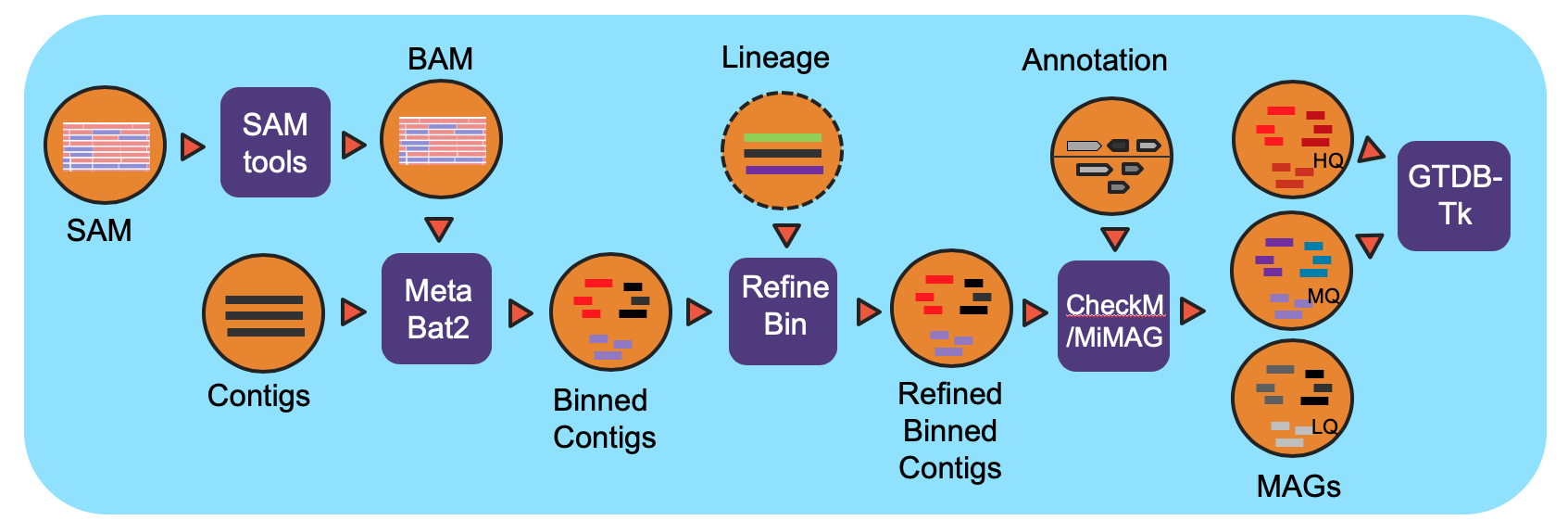
This workflow classifies contigs into bins and the resulting bins are refined using the functional annotation file. The bins are evaluated for completeness and contamination. The quality of the bins is determined and a lineage is assigned to each bin of high or medium quality.
Running the Workflow
Currently, this workflow is available in GitHub and can be run from the command line. (CLI instructions and requirements are found here.) Alternatively, this workflow can be run in NMDC EDGE.
Input
This workflow requires assembled contigs in a FASTA file, the read mapping file from the assembly (SAM or BAM), a functional annotation of the assembly in a GFF file.
Acceptable file formats: assembled contigs (.fasta, .fa, or .fna); read mapping to assembly (.sam.gz or .bam); Functional annotation (.gff)
Details
The workflow is based on IMG metagenome binning pipeline and has been modified specifically for the NMDC project. For all processed metagenomes, it classifies contigs into bins using MetaBat2. Next, the bins are refined using the functional Annotation file (GFF) from the Metagenome Annotation workflow and optional contig lineage information. The completeness of and the contamination present in the bins are evaluated by CheckM and bins are assigned a quality level (High Quality (HQ), Medium Quality (MQ), Low Quality (LQ)) based on MiMAG standards. In the end, GTDB-Tk is used to assign lineage for HQ and MQ bins. The required GTDB-Tk database is incorporated into NMDC EDGE. Full documentation can be found in ReadtheDocs.
Software Versions
Biopython v1.74
Sqlite
Pymysql
requests
samtools > v1.9 (License: MIT License)
Metabat2 v2.15
CheckM v1.1.2
GTDB-TK v1.2.0
FastANI v1.3
FastTree v2.1.10
Output
Multiple output files are provided by the workflow; the primary files are shown below. The full list of output files can be found in ReadtheDocs.
| Primary Output Files | Description |
|---|---|
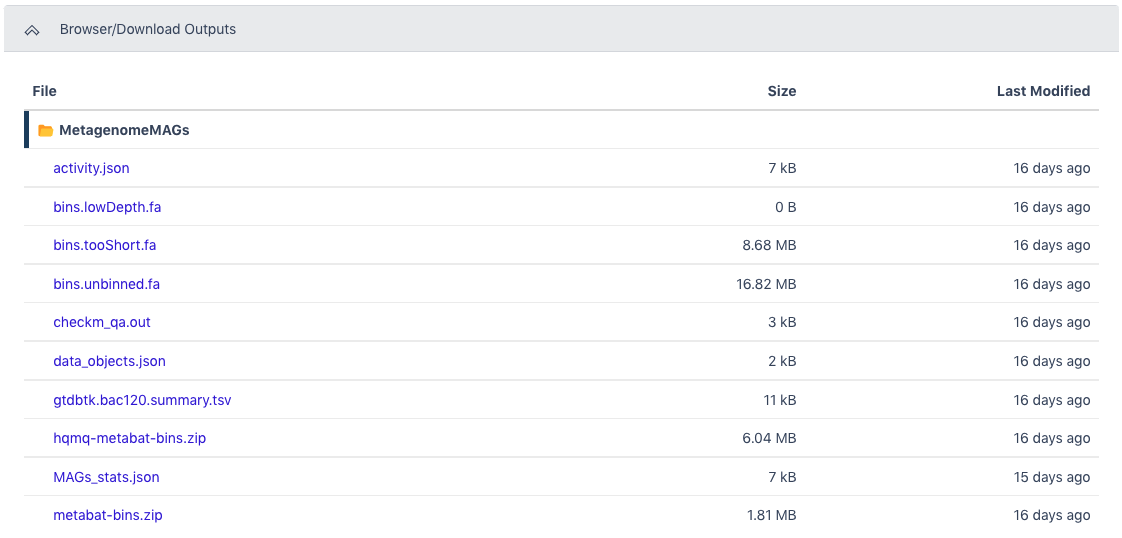
| hqmq-metabat-bins.zip | Bins of contigs rated high or medium quality with an assigned lineage |
Running the Metagenome Assembled Genomes (MAGs) Workflow in NMDC EDGE
Select a workflow
From the Metagenomics category in the left menu bar, select ‘Run a Single Workflow’.
Enter a unique project name with no spaces (underscores are fine).
A description is optional, but helpful.
Select ‘Metagenome MAGs’ from the dropdown menu under Workflow.
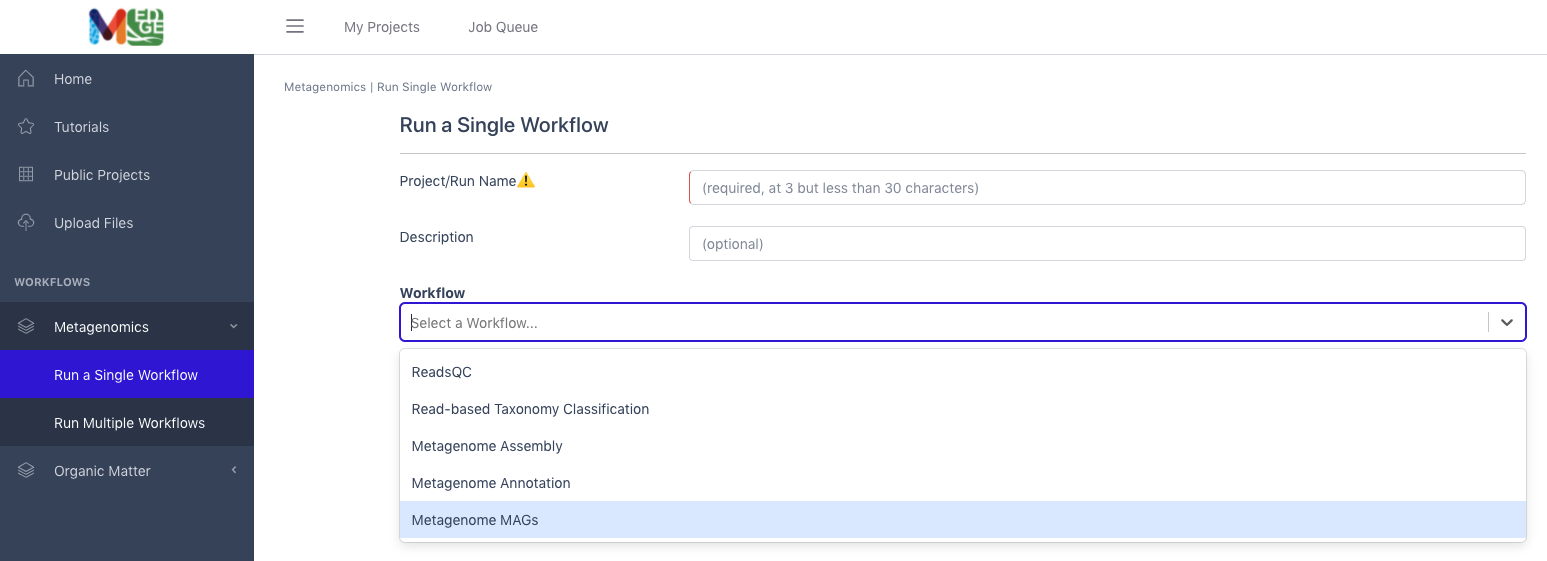
Input
Metagenome MAGs requires assembled contigs, the read mapping file of reads to assembled contigs, and a functional annotation file. The recommended input would be from the NMDC assembly and annotation workflows. Acceptable file formats: assembled contigs (.fasta, .fa, or .fna); read mapping to assembly (.sam.gz or .bam); functional annotation (.gff)
Click the button to the right of the blank for Input Contig File. A box called ‘Select a File’ will open to allow the user to find the desired file from a previously run assembly project, the public data folder, or a file uploaded by the user.
Click the button to the right of the blank for Input Sam/Bam File. A box called ‘Select a File’ will open to allow the user to find the read mapping file from a previously run assembly project, the public data folder, or a file uploaded by the user.
Click the button to the right of the blank for Input GFF File. A box called ‘Select a File’ will open to allow the user to find the desired file(s) from a previously run annotation project, the public data folder, or a file uploaded by the user.
Then click ‘Submit’.
Output
The General section of the output shows which workflow was run and the run time information.
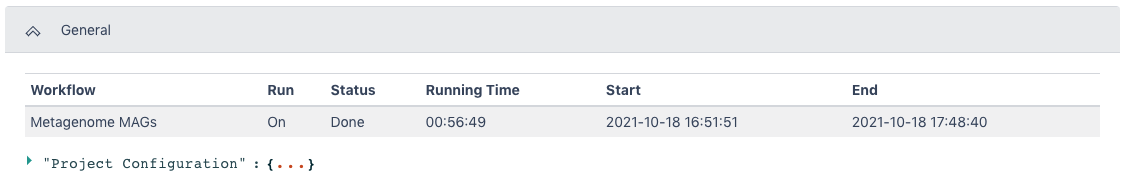
The Metagenome MAGs Result section provides a Summary section with information on binned and unbinned contigs. The MAGs section provides information such as the completeness of the genome, amount of contamination, and number of genes present on all bins determined to be high quality or medium quality.
The Browser/Download Output section provides output files available to download. The primary output file is the zipped file with all bins determined to be high quality or medium quality (hqmq-metabat-bins.zip).
Running multiple workflows or the full metagenomic pipeline with a single input
Metatranscriptomics Workflow
Overview
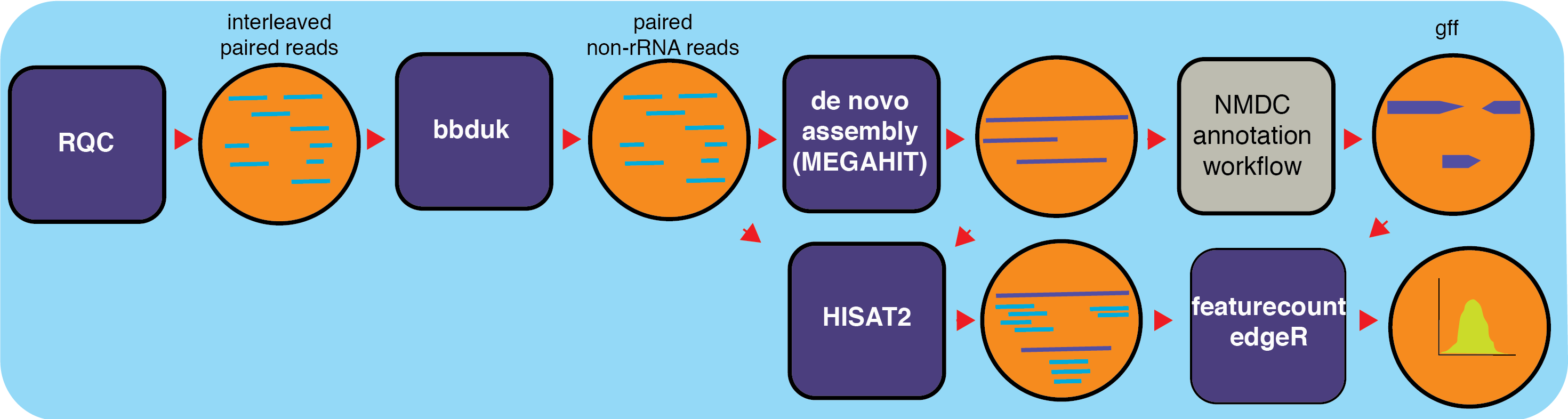
The metatranscriptome (metaT) workflow takes in raw metatranscriptome data, filters the data for quality, removes rRNA reads, then assembles and annotates the transcripts. The data is mapped back to the genomic features in the transcripts and RPKMs ((Reads Per Kilobase of transcript per Million mapped reads) are calculated for each feature in the functional annotation file.
Running the Workflow
Currently, this workflow can be run in NMDC EDGE or from the command line. (CLI instructions and requirements are found here.)
Input
Metatranscriptomics requires paired-end Illumina data as an interleaved file or as separate pairs of FASTQ files.
Acceptable file formats: .fastq, .fq, .fastq.gz, .fq.gz
Details
MetaT is a workflow designed to analyze metatranscriptomes, and this workflow builds upon other NMDC workflows for processing input sequencing data. The metatranscriptomics workflow takes in raw RNA sequencing data and quality filters the reads using the ReadsQC workflow. Then the MetaT workflow filters out ribosomal RNA reads (using the SILVA rRNA database) and separates interleaved files into separate pairs of files using bbduk (BBTools). After the filtering steps, the reads are assembled into transcripts using MEGAHIT and transcripts are annotated using the Metagenome Annotation NMDC Workflow which produces GFF functional annotation files. Features are counted with Subread’s featureCounts which assigns mapped reads to genomic features and generates RPKMs for each feature in a GFF file for sense and antisense reads.
Software Versions
BBTools v38.44
hisat2 v2.1
Python v3.7.6.
featureCounts v2.0.1
R v3.6.0
edgeR v3.28.1
pandas v1.0.5
gffutils v0.10.1
Output
The table below lists the primary output files. The main outputs are the assembled transcripts and annotated features file. Several annotation files are also available to download.
| Primary Output Files | Description |
|---|---|
| INPUT_NAME.contigs.fa | Assembled transcripts |
| rpkm_sorted_features.tsv | Feature table sorted by RPKM |
Running the Metatranscriptomics Workflow in NMDC EDGE
Select a workflow
From the Metatranscriptomics category in the left menu bar, select ‘Run a Single Workflow’.
Enter a unique project name with no spaces (underscores are fine).
A description is optional, but helpful.
Select ‘Metatranscriptome’ from the dropdown menu under Workflow.
Input
The metatranscriptome workflow requires paired-end Illumina data in FASTQ format as the input; the file can be interleaved and can be compressed. Acceptable file formats: .fastq, .fq, .fastq.gz, .fq.gz
The default setting is for the raw data to be in an interleaved format (paired reads interleaved into one file). If the raw data is paired reads in separate files (forward and reverse), click ‘No’.
Additional data files (of the same type–interleaved or separate) can be added with the button below.
Click the button to the right of the input blank to select the data file for the analysis. (If there are separate files, there will be two input blanks.) A ‘Select a File’ box will open to allow the user to find the desired file(s) from previously run projects, the public data folder, or files uploaded by the user.
Click ‘Submit’ when ready to run the workflow.
Output
The General section of the output shows which workflow was run, the run time information, and the Project Configuration
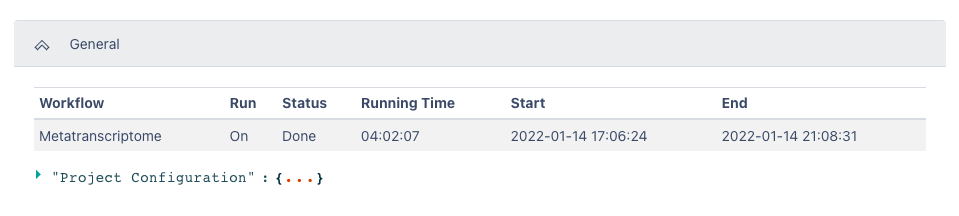
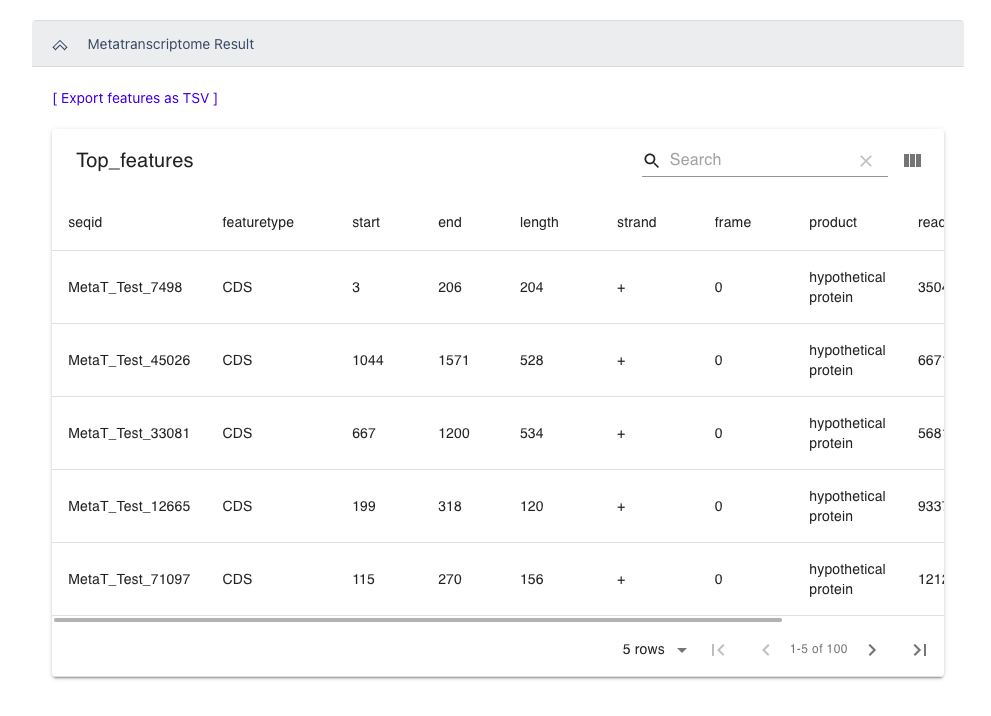
The Metatranscriptome Result section includes a table of the top 100 RPKM results from the overall metatranscriptome data file sorted by RPKM. Selecting the header of each column will sort this data by that column. This section also includes a button to quickly download a tsv file of all detected features in the input dataset for further analysis.
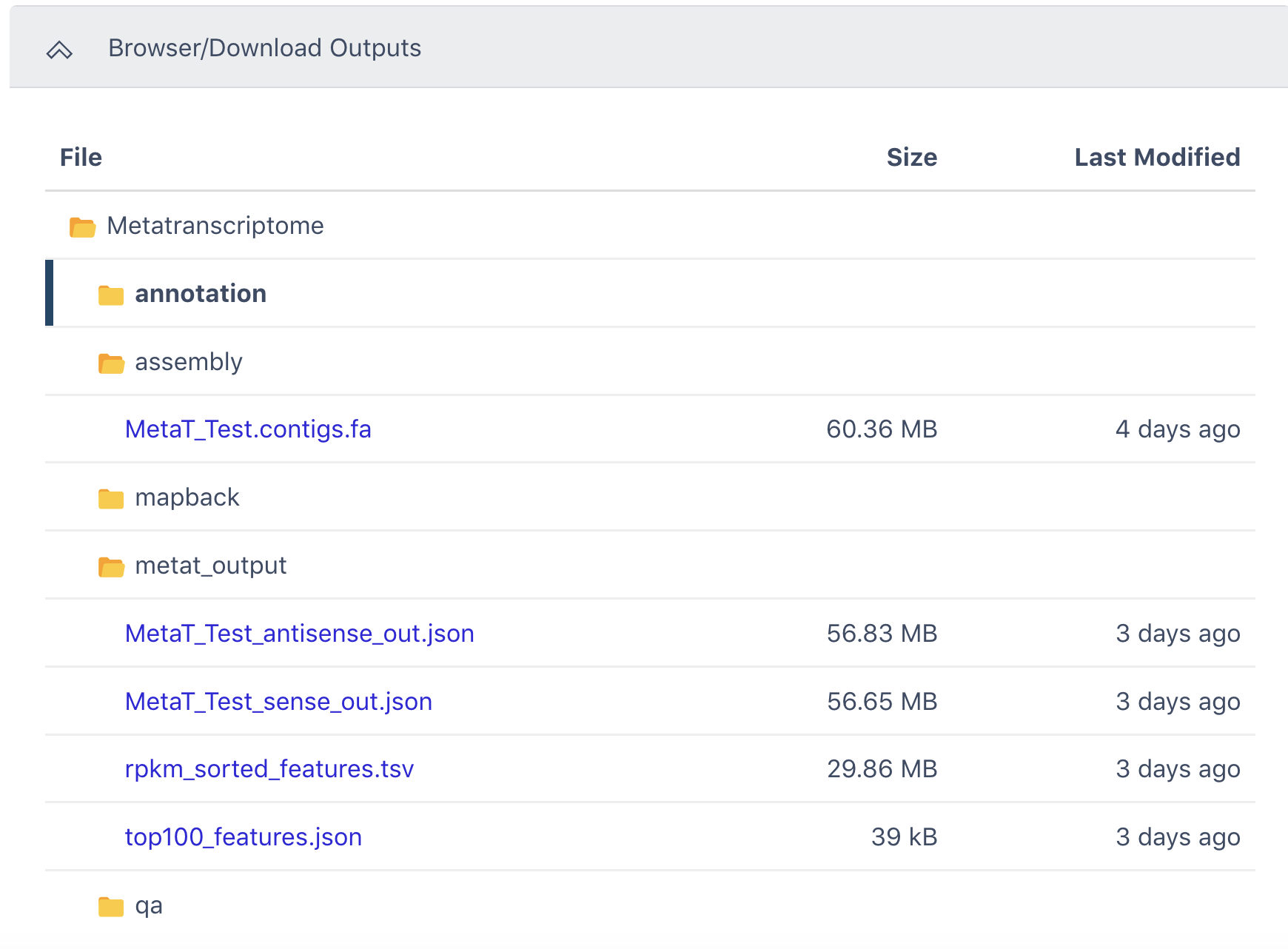
The Browser/Download Output section provides all output files available to download. The output contigs can be found in the assembly folder and the tsv file of all detected features sorted by RPKM is available under the metat_output folder.
Natural Organic Matter Workflow
Overview
This workflow takes FTICR mass spectrometry data collected from organic extracts to determine the molecular formulas of natural organic biomolecules in the input sample.
Running the Workflow
Currently, this workflow can be run in NMDC EDGE or from the command line. (CLI instructions and requirements are found here.)
Input
The input for this workflow is the output from a massSpec experiment (a massSpec list) which includes a minimum of two columns of data: a mass-to-charge ratio (m/z) and a signal intensity (Intensity) column for every feature in the analysis. A calibration file of molecular formula references is also required when running the workflow via command line. (This calibration file is built into NMDC EDGE.)
Acceptable file formats: .raw, .tsv, .csv, .xlsx
Details
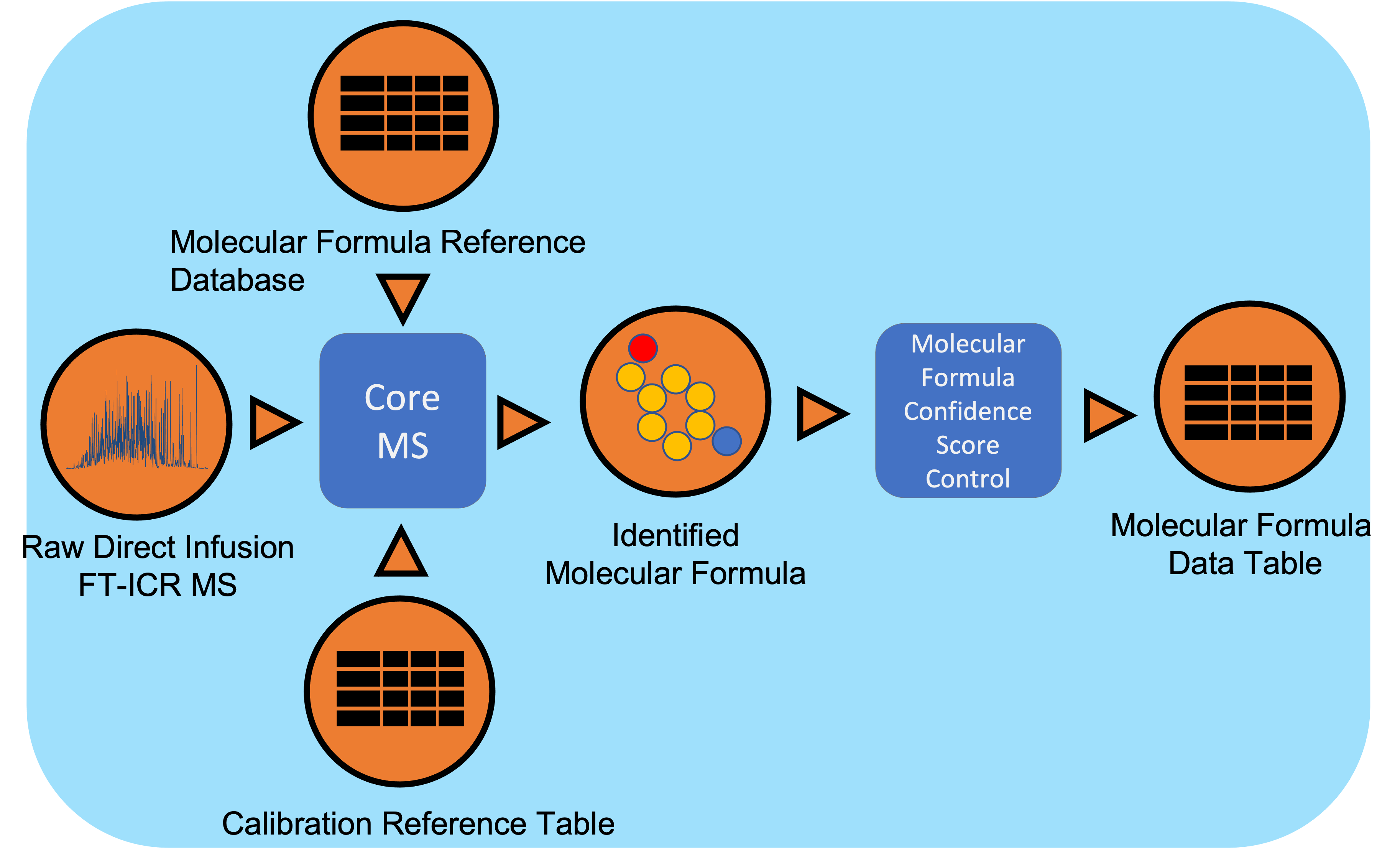
Direct Infusion Fourier Transform Ion Cyclotron Resonance mass spectrometry (DI FTICR-MS) data undergoes signal processing and molecular formula assignment leveraging EMSL’s CoreMS framework. Raw time domain data is transformed into the m/z domain using Fourier Transform and Ledford equation. Data is denoised followed by peak picking, recalibration using an external reference list of known compounds, and searched against a dynamically generated molecular formula library with a defined molecular search space. The confidence scores for all the molecular formula candidates are calculated based on the mass accuracy and fine isotopic structure, and the best candidate assigned as the highest score. This workflow will not work as reliably with Orbitrap mass spectrometry data.
Software Versions
CoreMS (2-clause BSD)
Click (BSD 3-Clause “New” or “Revised” License)
Output
The primary output file is the Molecular Formula Data Table (in a .csv file).
| Primary Output Files | Description |
|---|---|
| INPUT_NAME.csv | m/z, Peak height, Peak Area, Molecular Formula IDs, Confidence Score, etc. |
Running the Natural Organic Matter Workflow in NMDC EDGE
Select a workflow
From the Organic Matter category in the left menu bar, select ‘Run a Single Workflow’.
Enter a unique project name with no spaces (underscores are fine).
A description is optional, but helpful.
Select ‘EnviroMS’ from the dropdown menu under Workflow.
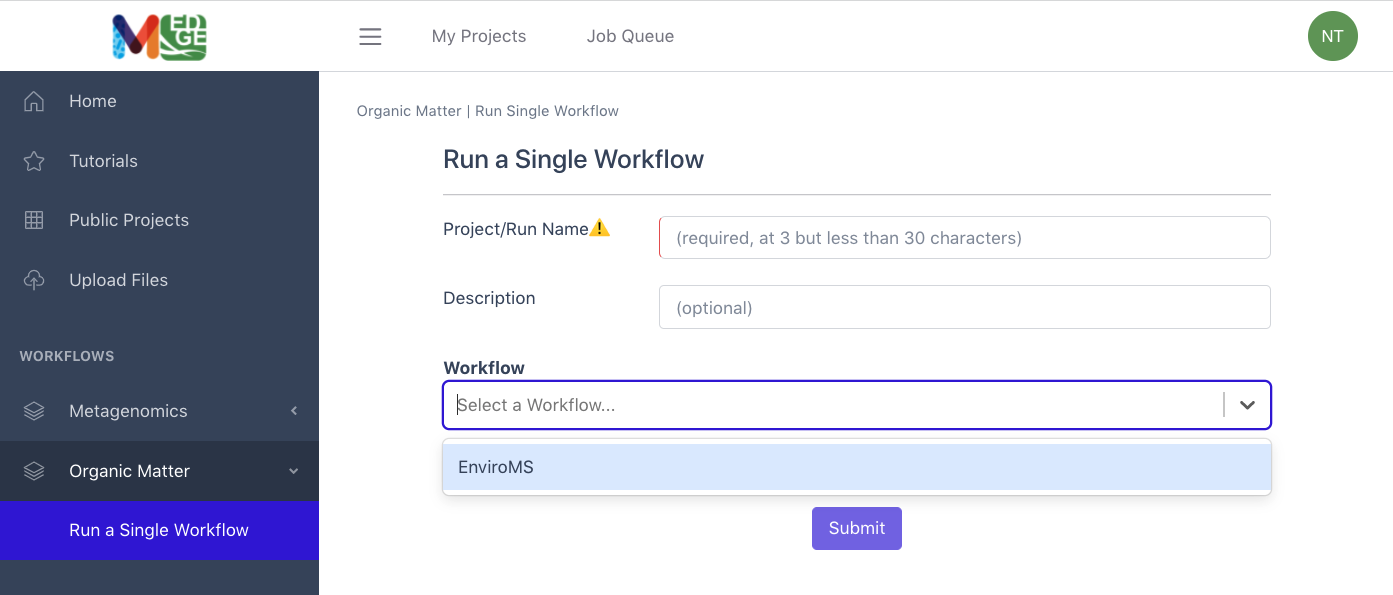
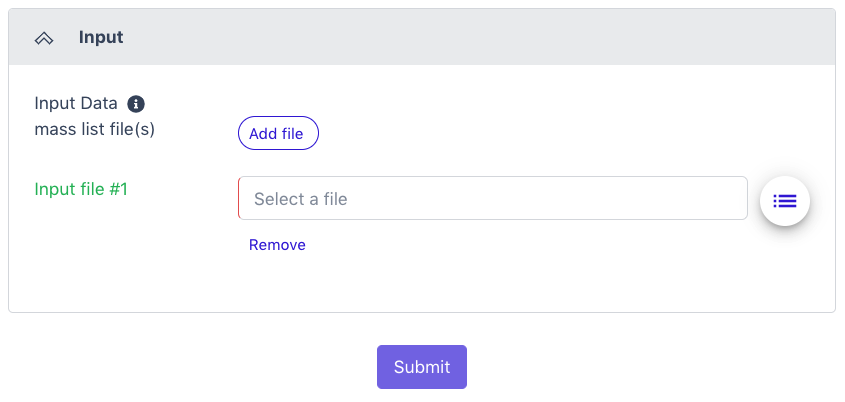
Input
The Natural Organic Matter workflow input is the output from a massSpec experiment (a massSpec list) with a minimum of two columns of data: a mass-to-charge ratio (m/z) and a signal intensity (Intensity) column for every feature in the analysis. Acceptable file formats: .tsv, .csv, .raw, .xlsx
Click the button to the right of the input blank for data to select the data file for the analysis. (If there are separate files, there will be two input blanks.) A box called ‘Select a File’ will open to allow the user to find the desired file(s) from the public data folder or files uploaded by the user.
Additional input files can be added by clicking the ‘Add file’ button to create additional input blanks.
Once all the input files have been selected, click ‘Submit’.
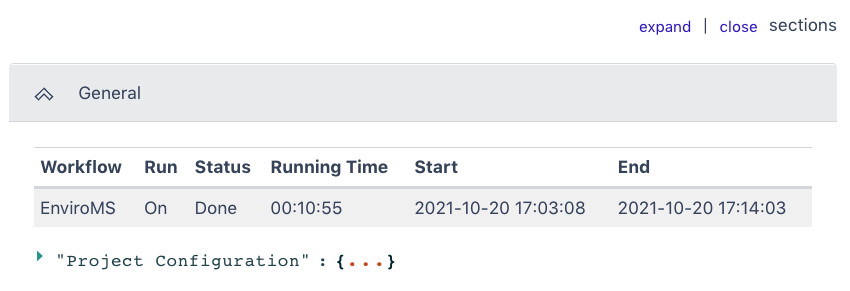
Output
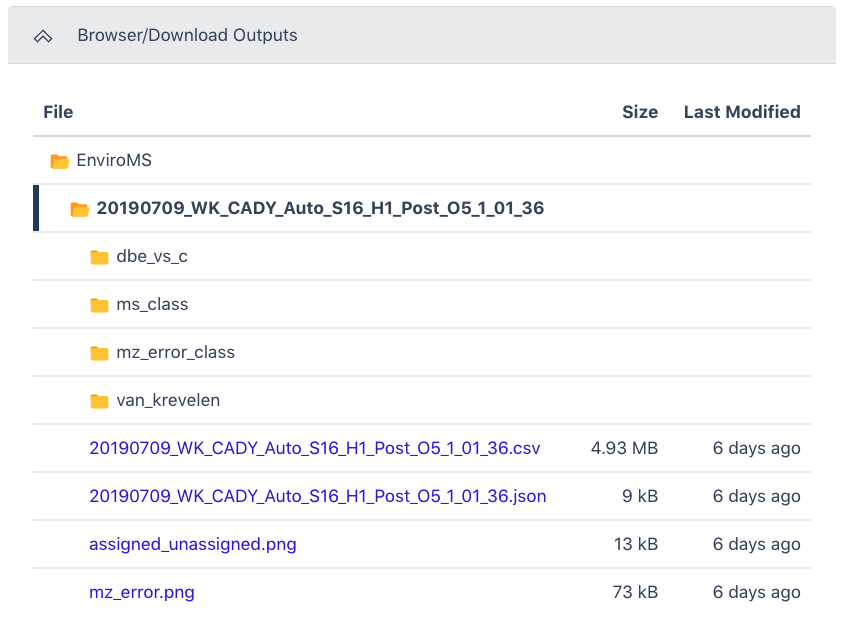
The General section of the output shows which workflow was run and the run time information. The Project Configuration can be seen by clicking the three dots in the bracket.
The Browser/Download Output section provides output files available to download. The primary output files are: the Molecular Formula Data-Table (.csv file) containing m/z measurements, Peak height, Peak Area, Molecular Formula Identification, Ion Type, and Confidence Score.