The NMDC Data Portal User Guide
Introduction
The pilot NMDC Data Portal (https://data.microbiomedata.org) provides a resource for consistently processed multi-omics data that is integrated to enable search, access, analysis, and download. Open-source bioinformatics workflows are used to process raw multi-omics data and produce interoperable and reusable annotated data from metagenome, metatranscriptome, metaproteome, metabolome, and natural organic matter characterizations. The NMDC Data Portal offers several search and navigation components, and data can be downloaded through the graphical user interface using an ORCiD authentication, with associated download metrics, or retrieved through available RESTful APIs. All multi-omics data are available under a Creative Commons 4.0 license, which enables public use with attribution, as outlined in the NMDC Data Use Policy (https://microbiomedata.org/nmdc-data-use-policy). This first iteration of the NMDC Data Portal was released in March 2021, and will continue to expand its data hostings and functionality on an ongoing.
There is a short video tutorial showing how to navigate the portal on Youtube (https://www.youtube.com/watch?v=KJQDrCnJRho).
User-Centered Design Process
The NMDC is a resource designed together with and for the scientific community. We have engaged in extensive user research through interviews and direct collaboration with the scientific community that have informed the design, development, and display of data through the NMDC Data Portal. This methodology (1) enables the scientific community to provide feedback, iterative and continuous improvement of our systems, and ensures that our systems enable a high level of scientific productivity. Feedback collected from the scientific community during early iterations of the Data Portal can be linked to the features and design directions found in the current release. Our community-centered design approach ensures that the NMDC can evolve with the needs of the microbiome research community, but will also be important for uncovering creative design solutions, clarifying expectations, reducing redesign, and perhaps most importantly, enabling shared ownership (2) of the NMDC. We hope that this inclusive approach will enable us to expand our engagements with the microbiome research community and the utility of the NMDC Data Portal.
Available Studies & Data
Data hostings include studies, biosamples, and 5 data types from a breadth of environmental microbiomes, spanning river sediments, subsurface shale carbon reservoirs, plant-microbe associations, and temperate and tropical soils. Specifics are as follows:
Studies
As the NMDC Data Portal is a pilot infrastructure, incoming projects for which study information and curated environmental metadata become available is first validated and loaded with a flag (Omics data coming soon) before processed instrumentation data is integrated into the portal.
Standards
The NMDC team works closely with several standards groups and organizations. We have adopted the Genomic Standards Consortium (GSC) Minimum Information about any (x) Sequence (MIxS) templates (3). This provides a standard data dictionary of sample descriptors (e.g., location, biome, altitude, depth) organized into seventeen environmental packages (https://www.gensc.org/pages/standards-intro.html) for sequence data. The NMDC team has mapped fields used to describe samples in the GOLD database to MIxS version 6.1 elements. In addition, we are adopting the MIxS standards for sequence data types (e.g., sequencing method, pcr primers and conditions, etc.), and are leveraging standards and controlled vocabularies developed by the Proteomics Standards Initiative (4), the National Cancer Institute’s Proteomic Data Commons (https://pdc.cancer.gov/data-dictionary/dictionary.html), and the Metabolomics Standards Initiative (5) for mass spectrometry data types (e.g., ionization mode, mass resolution, scan rate, etc.).
MIxS environmental packages
The GSC has developed standards for describing genomic and metagenomic sequences, and the environment from which a biological sample originates. These “Minimum Information about any (x) Sequence” (MIxS) packages provides standardized sample descriptors (e.g., location, environment, elevation, altitude, depth, etc.) for 17 different sample environments.
Environment Ontology (EnvO)
EnvO is a community-led ontology that represents environmental entities such as biomes, environmental features, and environmental materials. These EnvO entities are the recommended values for several of the mandatory terms in the MIxS packages, often referred to as the “MIxS triad”.
Genomes OnLine Database (GOLD)
GOLD is an open-access repository of genome, metagenome, and metatranscriptome sequencing projects with their associated metadata. Biosamples (defined as the physical material collected from an environment) are described using a five-level ecosystem classification path that goes from ecosystem down to the type of environmental material that describes the sample.
Omics Data
A suite of omics processing data can be generated from available biosamples, and the value of associating these data through a common sample source enables researchers to probe function. The NMDC data schema offers an approach to link omics processing runs to their source biosample (for example, multiple organic matter characterizations can be generated from a single sample through extraction with various solvents, eg, chloroform, methanol, and water fractionation). Below outlines the various omics data currently available through the portal.
Metagenomes.
Illumina-sequenced shotgun metagenome data undergo pre-processing, error correction, assembly, structural and functional annotation, and binning leveraging the JGI’s production pipelines (6), along with an additional read-based taxonomic analysis component. Standardized outputs from the read QC, read-based analysis, assembly, annotation, and binning are available for search and download on the NMDC Data Portal.
Metatranscriptomes.
Illumina-sequenced shotgun reads from cDNA library undergo pre-processing and error correction in the same way as described above in the metagenome workflow with additional steps to filter ribosomal reads. High-quality reads are then assembled into transcripts using MEGAHIT (7), annotated using the annotation module described in the metagenome workflow, and the high-quality reads are mapped back to the annotated transcripts using HISAT2 (8) and then processed to calculate the number of reads mapped per feature using FeatureCount (9) and RPKM calculations per feature using edgeR (10). Results from read QC, assembly, and annotation are available for search and download for metatranscriptomes on the NMDC Data Portal.
Metaproteomes.
Data-dependent mass spectrometry raw data files are first converted to mzML, using MSConvert (11). Peptide identification is achieved using MSGF+ (12) and the associated metagenomic information in the FASTA file. Peptide identification false discovery rate is controlled using a decoy database approach. Intensity information is extracted using MASIC (13) and combined with protein information. Protein annotation information is obtained from the associated metagenome annotation output. Standardized outputs for quality control, and peptide and protein-level quantitative data are available for search and download for metaproteomes on the NMDC Data Portal.
Metabolomes.
The gas chromatography-mass spectrometry (GC-MS) based metabolomics workflow (metaMS) developed by leveraging EMSL’s CoreMS mass spectrometry software framework allows target and semi-target data analysis of metabolomics data (14). The raw data is parsed into coreMS data structure and undergoes all the steps of signal processing (signal noise reduction, m/z based chromatogram peak deconvolution, abundance threshold calculation, peak picking) and molecular identification, including the molecular search using a metabolites standard compound library, spectral similarity calculation, and similarity score calculation (15), all in a single step. The putative metabolite annotation data is available to download for metabolomes on the NMDC Data Portal. Data dependent LC-MS based workflows are currently under development. Additionally, it should be noted that all available data derives from exploratory, untargeted analysis and is semi-quantitative.
Natural Organic Matter Characterization (NOM).
Direct Infusion Fourier Transform mass spectrometry (DI FT-MS) data undergoes signal processing and molecular formula assignment leveraging EMSL’s CoreMS framework (14). Raw time domain data is transformed into the m/z domain using Fourier Transform and Ledford equation (16). Data is denoised followed by peak picking, recalibration using an external reference list of known compounds, and searched against a dynamically generated molecular formula library with a defined molecular search space. The confidence scores for all the molecular formula candidates are calculated based on the mass accuracy and fine isotopic structure, and the best candidate assigned as the highest score. The molecular formula characterization table is available to download for natural organic matter characterizations on the NMDC Data Portal.
Portal Functionality
#Faceted search and access
Search by investigator name
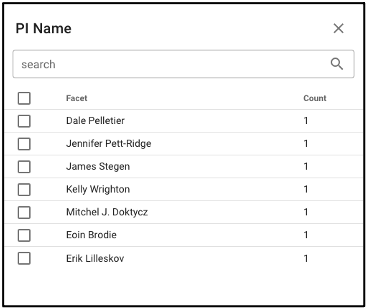
NMDC-linked data can be filtered by the associated principal investigator by selecting ‘PI Name’ from the left query term bar. This selection will display studies and samples associated with that PI, and selecting the arrow on the right side of the study name will open up more information about that study and that principal investigator.
Search by omics processing information
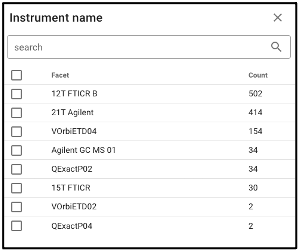
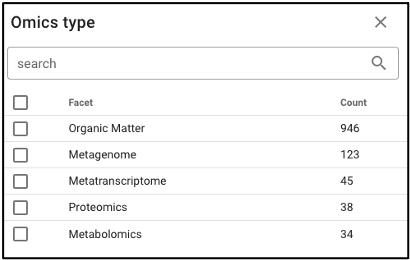

Samples can be queried by various omics processing information terms including instrument name, omics type (processing runs sorted by omics type can also be queried using the bar plot on the main portal page), and processing institution.
Search by KEGG Orthology (KO)
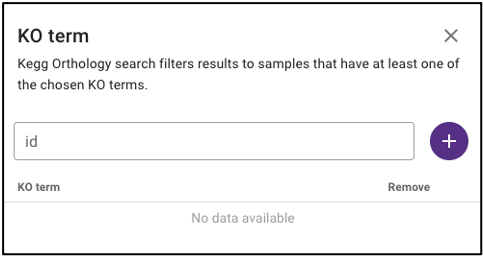
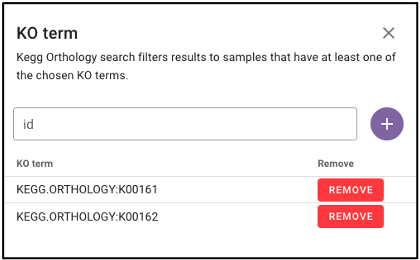
Under ‘Function’ on the query term bar, users are able to search by KEGG Orthology (KO) terms to limit the query to samples with datasets that include at least one of the listed KO terms. Users may list multiple KO terms, but it is important to note that adding multiple terms will limit the search to datasets that include at least one of those KO terms, not all of the added terms.
Search by environmental descriptors
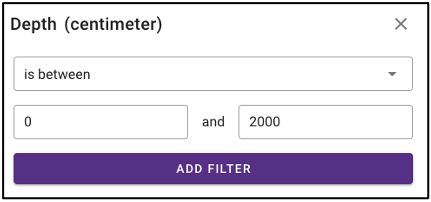
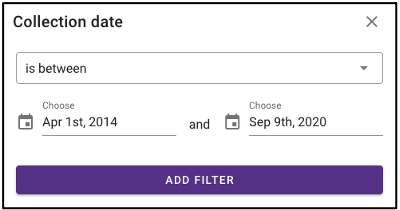
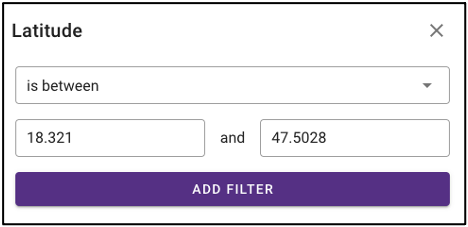
The query term bar also includes several environmental descriptor filtering fields of where the samples were isolated from. Users can filter by sample isolation depth, collection date, latitude and longitude (can also filter by latitude and longitude using the interactive map on the omics main page), as well as geographic location name.
Search by ecosystem classifications

Samples can also be queried by ecosystem classifications using GOLD and/or ENVO terms. Selecting GOLD classification in the query term bar opens up a hierarchy that can be navigated through to select ecosystem classification(s) of interest. Users can select everything under a certain classification at any point, or can continue navigating to more specific classifications. The Sankey diagram on the ‘Environment’ page provides an interactive visualization of the GOLD classification system.
Similarly, ENVO terms can be used to query the portal, and these are broken down into environmental biome, feature, and material categories. ENVO is another effective classification system that can be used to describe environments where samples were collected from.
Interactive visualizations
Omics Page
Barplot
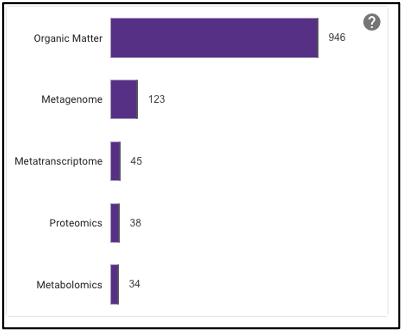
The barplot on the omics page displays the number of omics processing runs (not number of samples) for each data type available: organic matter, metagenomic, metatranscriptomic, proteomic, and metabolomic. Selecting the bar of a data type will limit the search to just that data type.
Geographic map
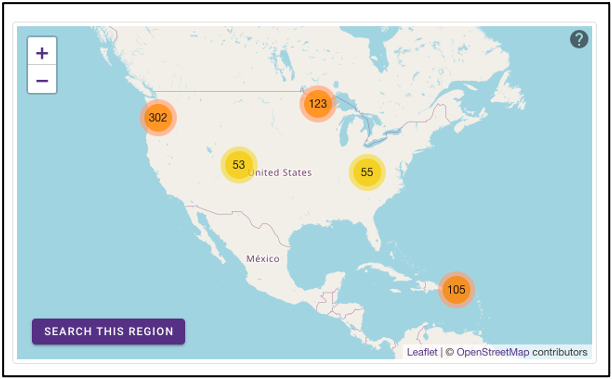
The geographic map on the omics page allows for samples to be queried by the geographic location from which they were isolated. The map displays the geographical location (latitude, longitude) of the sample collection sites as clusters with colors corresponding to the number of samples from that area. The map can be zoomed in and out of, and clusters can be selected to focus on that specific area. After zooming and moving around the map to a region of interest, selecting the ‘Search this region’ button will limit the search to the current map bounds.
Temporal slider
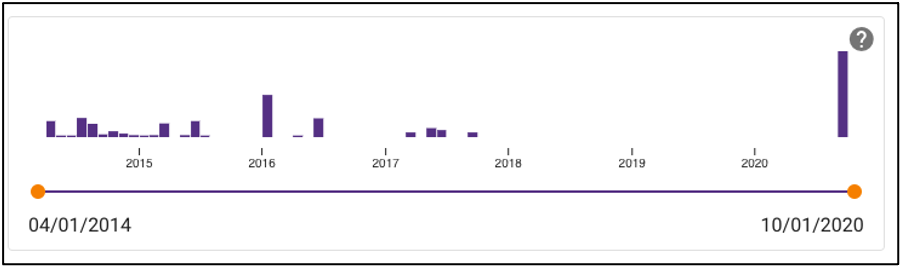
Samples can also be queried by a sample collection date range by dragging the dots below the temporal slider on the omics page. Sample collection dates are grouped by month.
Upset plot
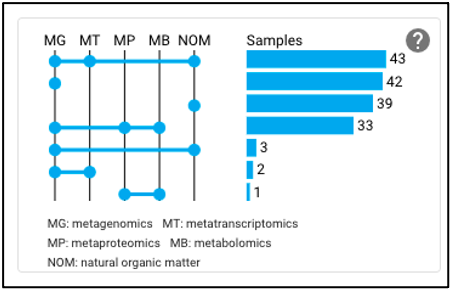
The upset plot on the omics page displays the number of samples that have various combinations of associated omics data. The axis at the top of the plot refers to the different omics types (MG: metagenomic, MT: metatranscriptomic, MP: metaproteomic, MB: metabolomic, NOM: natural organic matter) and the dots and lines in the graph below represent the combinations of the omics data types. The numbers and bars on the right side represent the number of samples searchable in the NMDC data portal with each corresponding combination of omics data types. This plot will update as query terms are added.
Environment Page
Sankey diagram
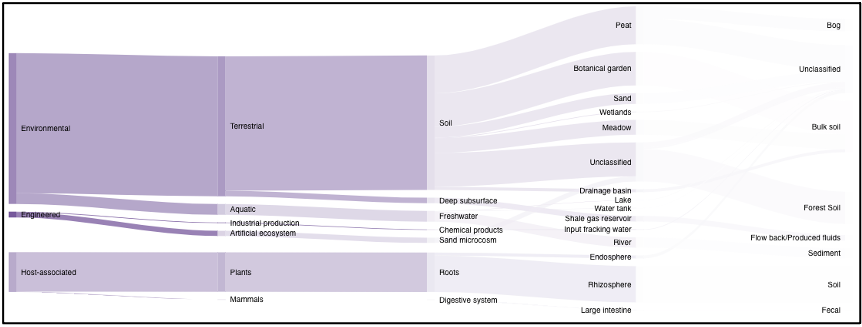
On the environment page, the Sankey diagram displays the environments that NMDC-linked samples were isolated from. This visualization is based on the GOLD ecosystem classification path, and the diagram is fully interactive, so environments of interest can be chosen at descending levels of specificity. This will then limit your search to samples that came from that selected environment.
Download
Individual file
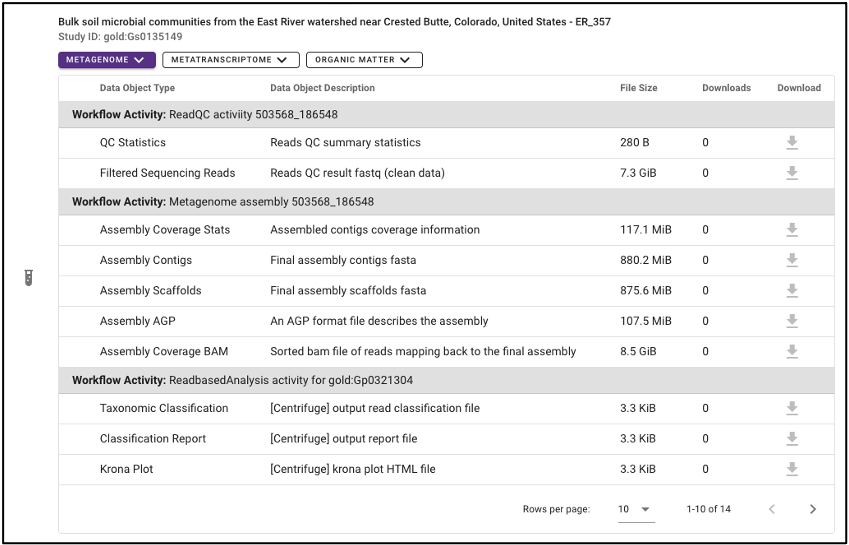
Various output data files are available from samples findable through the NMDC that have been run through the NMDC standardized workflows. Output files from each omic type are sorted by the specific workflow (e.g. Metagenome Assembly, Annotation) that was run and are each available for download when the sample of interest is selected. Users must log in with an ORCID account before downloading data.
Bulk download
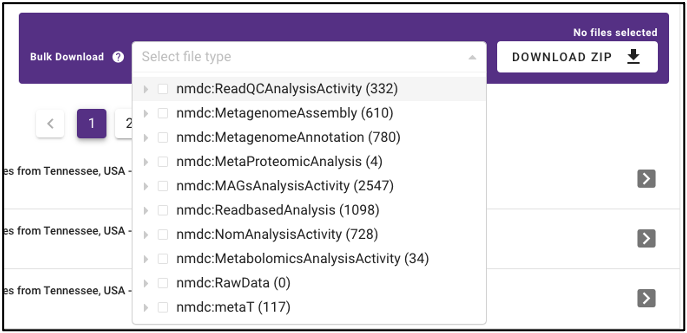
In addition to the ability to download single output files from samples run through the NMDC standardized workflows, the NMDC portal allows users to perform bulk downloads on workflow output files. Once samples of interest are down-selected through query terms, output files from each NMDC standardized workflow run on those samples are available as bulk downloads. Users must be logged in with an ORCID account before downloading data.
References
Abras C, Maloney-Krichmar, D., Preece, J. 2004. User-Centered Design. _In _Bainbridge W (ed), Encyclopedia of Human-Computer Interaction. Sage Publications, Thousand Oaks.
Preece J, Rogers, Y., & Sharp, H. 2002. Interaction design: Beyond human-computer interaction. John Wiley & Sons, New York, NY.
Yilmaz P, Kottmann R, Field D, Knight R, Cole JR, Amaral-Zettler L, Gilbert JA, Karsch-Mizrachi I, Johnston A, Cochrane G, Vaughan R, Hunter C, Park J, Morrison N, Rocca-Serra P, Sterk P, Arumugam M, Bailey M, Baumgartner L, Birren BW, Blaser MJ, Bonazzi V, Booth T, Bork P, Bushman FD, Buttigieg PL, Chain PSG, Charlson E, Costello EK, Huot-Creasy H, Dawyndt P, DeSantis T, Fierer N, Fuhrman JA, Gallery RE, Gevers D, Gibbs RA, Gil IS, Gonzalez A, Gordon JI, Guralnick R, Hankeln W, Highlander S, Hugenholtz P, Jansson J, Kau AL, Kelley ST, Kennedy J, Knights D, Koren O, et al. 2011. Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. _Nature Biotechnol. _29:415-420.
Taylor CF, Paton NW, Lilley KS, Binz P-A, Julian RK, Jones AR, Zhu W, Apweiler R, Aebersold R, Deutsch EW, Dunn MJ, Heck AJR, Leitner A, Macht M, Mann M, Martens L, Neubert TA, Patterson SD, Ping P, Seymour SL, Souda P, Tsugita A, Vandekerckhove J, Vondriska TM, Whitelegge JP, Wilkins MR, Xenarios I, Yates JR, Hermjakob H. 2007. The minimum information about a proteomics experiment (MIAPE). _Nature Biotechnol. _25:887-893.
Sansone S-A, Fan T, Goodacre R, Griffin JL, Hardy NW, Kaddurah-Daouk R, Kristal BS, Lindon J, Mendes P, Morrison N, Nikolau B, Robertson D, Sumner LW, Taylor C, van der Werf M, van Ommen B, Fiehn O, Members MSIB. 2007. The Metabolomics Standards Initiative. _Nature Biotechnol. _25:846-848.
Clum A, Huntemann M, Bushnell B, Foster B, Foster B, Roux S, Hajek PP, Varghese N, Mukherjee S, Reddy TBK, Daum C, Yoshinaga Y, O’Malley R, Seshadri R, Kyrpides NC, Eloe-Fadrosh EA, Chen I-MA, Copeland A, Ivanova NN, Segata N. 2021. DOE JGI Metagenome Workflow. _mSystems _6:e00804-20.
Li D, Liu C-M, Luo R, Sadakane K, Lam T-W. 2015. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. _Bioinformatics _31:1674-1676.
Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. 2019. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. _Nature Biotechnol. _37:907-915.
Liao Y, Smyth GK, Shi W. 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. _Bioinformatics _30:923-30.
Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. _Bioinformatics _26:139-140.
Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, Gatto L, Fischer B, Pratt B, Egertson J, Hoff K, Kessner D, Tasman N, Shulman N, Frewen B, Baker TA, Brusniak MY, Paulse C, Creasy D, Flashner L, Kani K, Moulding C, Seymour SL, Nuwaysir LM, Lefebvre B, Kuhlmann F, Roark J, Rainer P, Detlev S, Hemenway T, Huhmer A, Langridge J, Connolly B, Chadick T, Holly K, Eckels J, Deutsch EW, Moritz RL, Katz JE, Agus DB, MacCoss M, Tabb DL, Mallick P. 2012. A cross-platform toolkit for mass spectrometry and proteomics. _Nature Biotechnol. _30:918-20.
Kim S, Gupta N, Pevzner PA. 2008. Spectral Probabilities and Generating Functions of Tandem Mass Spectra: A Strike against Decoy Databases. _J Proteome Res. _7:3354-3363.
Monroe ME, Shaw JL, Daly DS, Adkins JN, Smith RD. 2008. MASIC: A software program for fast quantitation and flexible visualization of chromatographic profiles from detected LC– MS(/MS) features. _Comp. Biol. Chemistry _32:215-217.
Corilo YE, Kew WR, McCue LA. 2021. EMSL-Computing/CoreMS: CoreMS 1.0.0 (v1.0.0). Zenodo. 10.5281/zenodo.4641552.
Hiller K, Hangebrauk J, Jäger C, Spura J, Schreiber K, Schomburg D. 2009. MetaboliteDetector: comprehensive analysis tool for targeted and nontargeted GC/MS based metabolome analysis. _Anal Chem _81:3429-39.
Marshall AG, Hendrickson CL, Jackson GS. 1998. Fourier transform ion cyclotron resonance mass spectrometry: a primer. _Mass Spectrom Rev _17:1-35.